
VDUP1 Deficiency Promotes the Severity of DSS-Induced Colitis in Mice by Inducing Macrophage Infiltration
 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
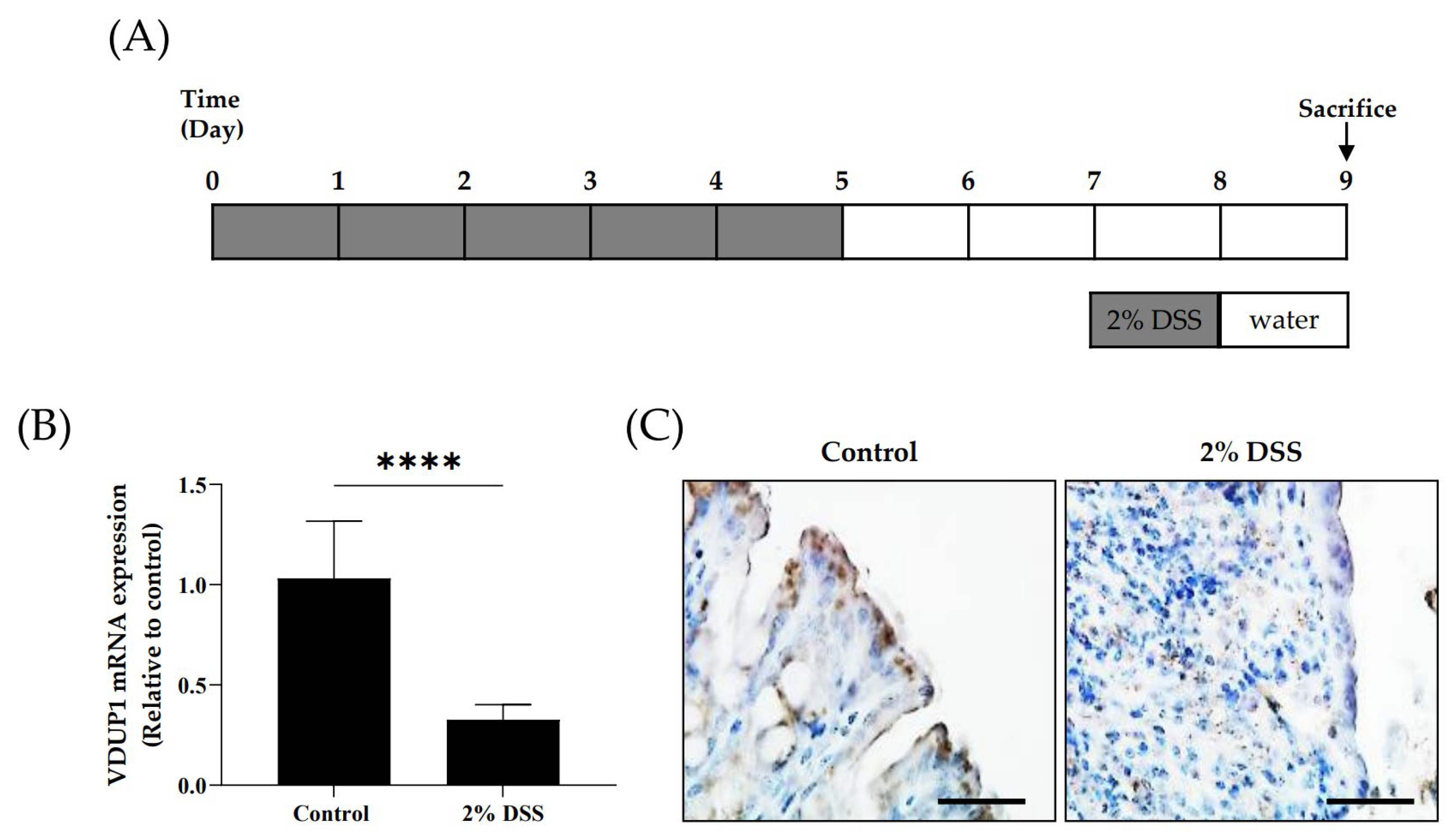
2.1. The Expression of VDUP1 Was Reduced in Experimental Colitis
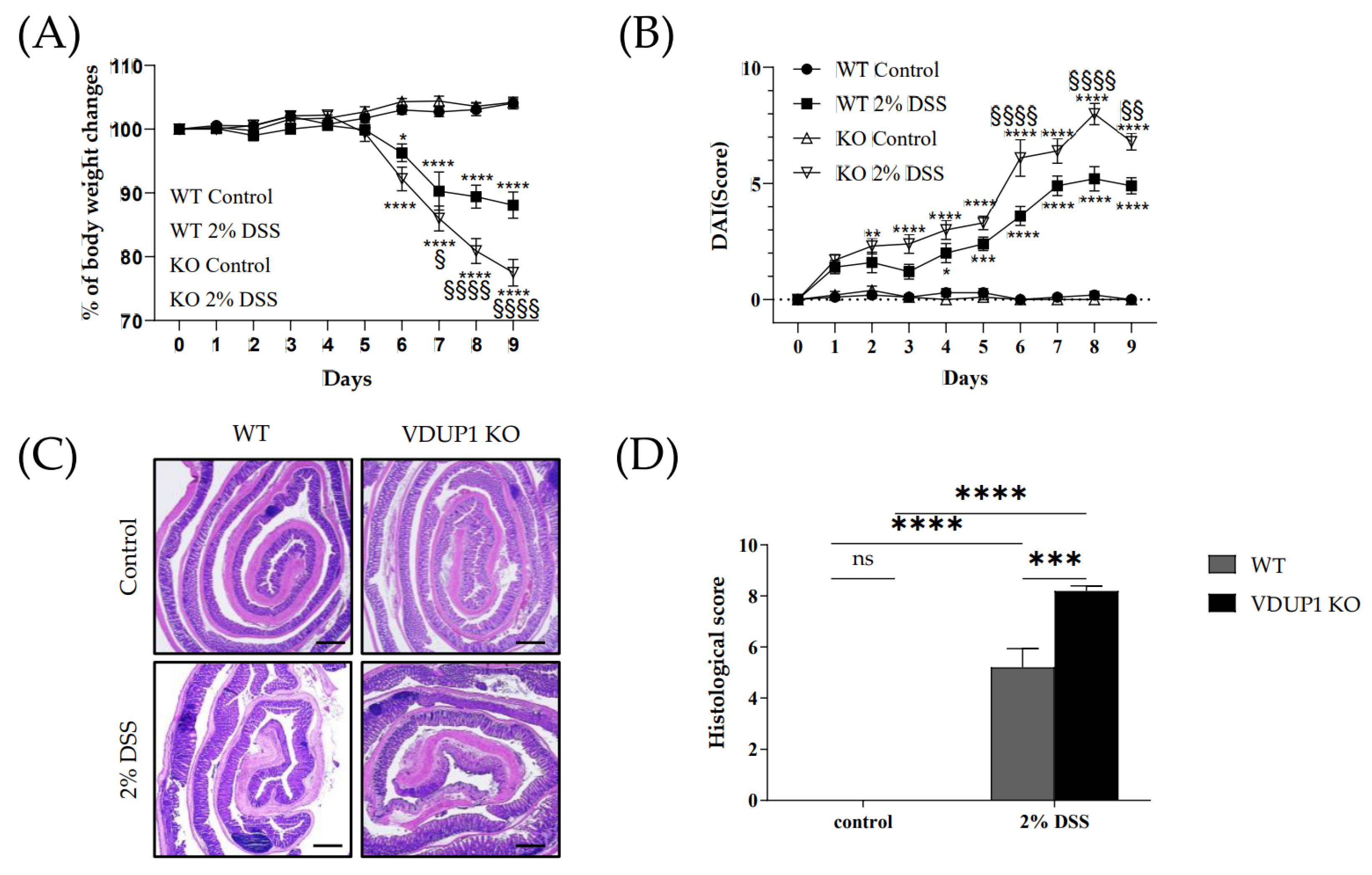
2.2. VDUP1 Deficiency Exacerbated the Severity of DSS-Induced Colitis
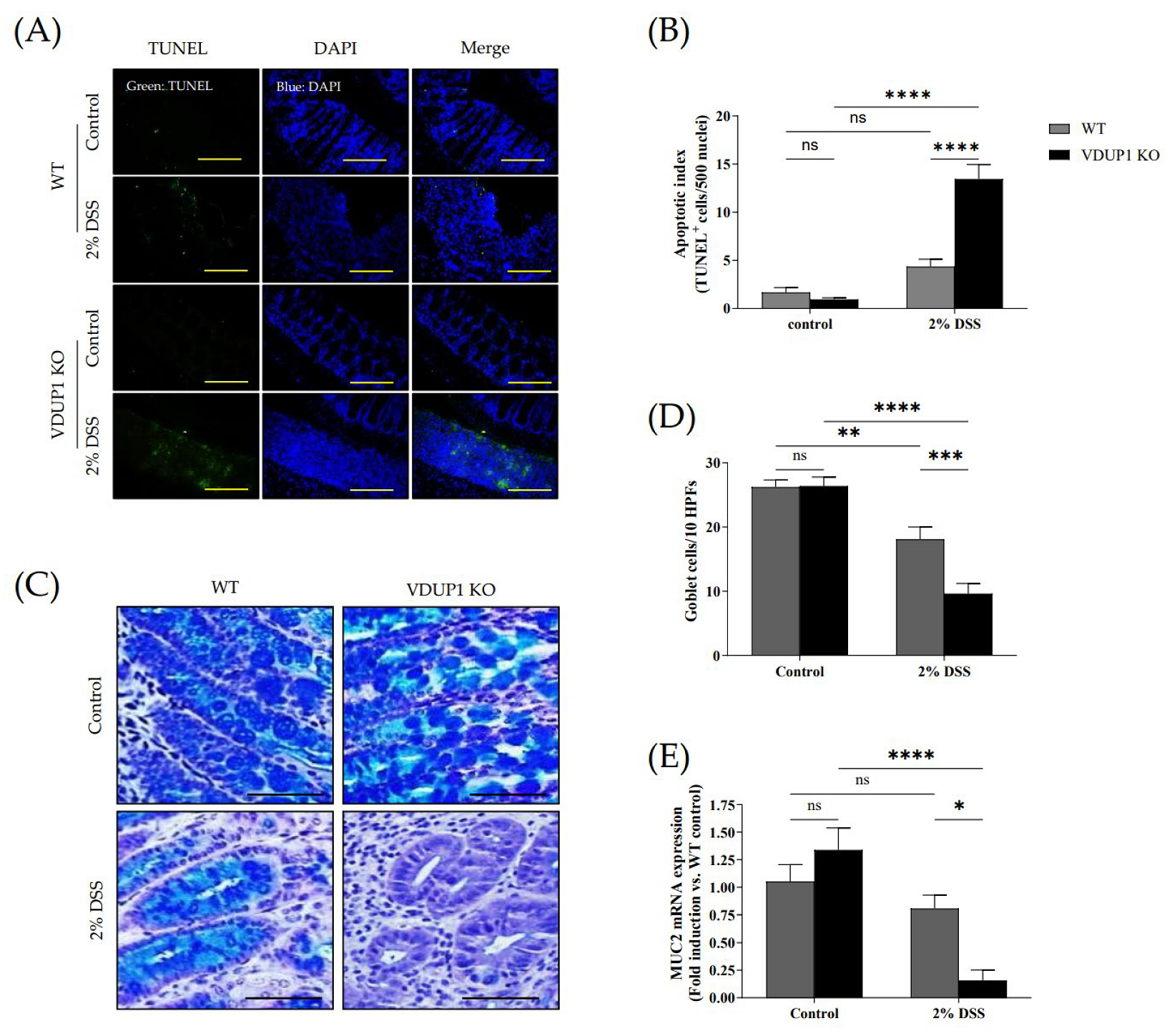
2.3. VDUP1 Deficiency Accelerated Colonic Tissue Damage in Experimental Colitis
2.4. VDUP1 Deficiency Induced Inflammatory Cytokine Expression in Experimental Colitis
2.5. VDUP1 Deficiency Activated NF-κB p65 in Experimental Colitis
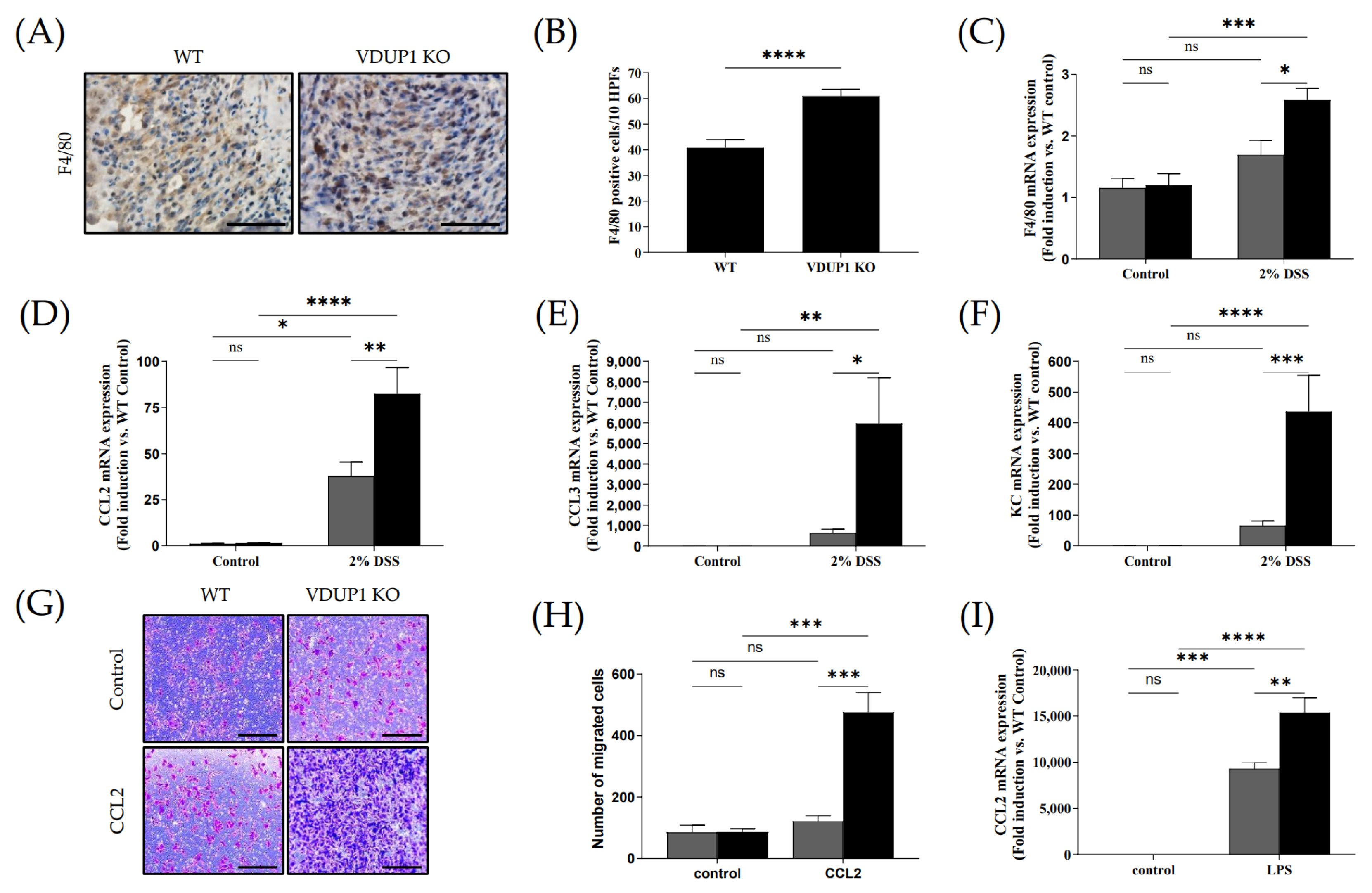
2.6. VDUP1 Deficiency Promoted Macrophage Chemotaxis to the Site of Inflammation
3. Discussion
4. Materials and Methods
4.1. Reagents and Animals
4.2. DSS-Induced Colitis
4.3. RNA Isolation and Quantification of mRNA Expression
4.4. Colon Histology, Immunohistochemistry, Immunofluorescence Analysis, and TUNEL Assay
4.5. AB-PAS Staining
4.6. Primary Cell Culture and Chemotaxis Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Head, K.A.; Jurenka, J.S. Inflammatory bowel disease Part 1: Ulcerative colitis-pathophysiology and conventional and alternative treatment options. Altern. Med. Rev. 2003, 8, 247–283. [Google Scholar] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Primers 2020, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Famularo, G.; Trinchieri, V.; De Simone, C. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 1982–1984. [Google Scholar]
- Qiu, W.; Wu, B.; Wang, X.; Buchanan, M.E.; Regueiro, M.D.; Hartman, D.J.; Schoen, R.E.; Yu, J.; Zhang, L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J. Clin. Investig. 2011, 121, 1722–1732. [Google Scholar] [CrossRef]
- Kiesslich, R.; Duckworth, C.A.; Moussata, D.; Gloeckner, A.; Lim, L.G.; Goetz, M.; Pritchard, D.M.; Galle, P.R.; Neurath, M.F.; Watson, A.J. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut 2012, 61, 1146–1153. [Google Scholar] [CrossRef]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef]
- Tatiya-Aphiradee, N.; Chatuphonprasert, W.; Jarukamjorn, K. Immune response and inflammatory pathway of ulcerative colitis. J. Basic. Clin. Physiol. Pharmacol. 2018, 30, 1–10. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Gelbmann, C.M.; Leeb, S.N.; Vogl, D.; Maendel, M.; Herfarth, H.; Scholmerich, J.; Falk, W.; Rogler, G. Inducible CD40 expression mediates NFkappaB activation and cytokine secretion in human colonic fibroblasts. Gut 2003, 52, 1448–1456. [Google Scholar] [CrossRef]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-kappaB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Fuss, I.; Schurmann, G.; Pettersson, S.; Arnold, K.; Muller-Lobeck, H.; Strober, W.; Herfarth, C.; Buschenfelde, K.H. Cytokine gene transcription by NF-kappa B family members in patients with inflammatory bowel disease. Ann. N. Y. Acad. Sci. 1998, 859, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Brand, K.; Vogl, D.; Page, S.; Hofmeister, R.; Andus, T.; Knuechel, R.; Baeuerle, P.A.; Scholmerich, J.; Gross, V. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology 1998, 115, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ding, S.; Jiang, H.; Liu, G. Roles of Macrophages in the Development and Treatment of Gut Inflammation. Front. Cell Dev. Biol. 2021, 9, 625423. [Google Scholar] [CrossRef]
- Lu, P.D.; Zhao, Y.H. Targeting NF-kappaB pathway for treating ulcerative colitis: Comprehensive regulatory characteristics of Chinese medicines. Chin. Med. 2020, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, D.; Zhou, C.; Cao, X.; He, J. Correlation of Macrophages with Inflammatory Reaction in Ulcerative Colitis and Influence of Curcumin on Macrophage Chemotaxis. Altern. Ther. Health Med. 2023, 29, 97–103. [Google Scholar]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef]
- Shalev, A.; Pise-Masison, C.A.; Radonovich, M.; Hoffmann, S.C.; Hirshberg, B.; Brady, J.N.; Harlan, D.M. Oligonucleotide microarray analysis of intact human pancreatic islets: Identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology 2002, 143, 3695–3698. [Google Scholar] [CrossRef]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef]
- Chen, J.; Jing, G.; Xu, G.; Shalev, A. Thioredoxin-interacting protein stimulates its own expression via a positive feedback loop. Mol. Endocrinol. 2014, 28, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef]
- Amin, F.M.; Abdelaziz, R.R.; Hamed, M.F.; Nader, M.A.; Shehatou, G.S.G. Dimethyl fumarate ameliorates diabetes-associated vascular complications through ROS-TXNIP-NLRP3 inflammasome pathway. Life Sci. 2020, 256, 117887. [Google Scholar] [CrossRef] [PubMed]
- Riahi, Y.; Kaiser, N.; Cohen, G.; Abd-Elrahman, I.; Blum, G.; Shapira, O.M.; Koler, T.; Simionescu, M.; Sima, A.V.; Zarkovic, N.; et al. Foam cell-derived 4-hydroxynonenal induces endothelial cell senescence in a TXNIP-dependent manner. J. Cell. Mol. Med. 2015, 19, 1887–1899. [Google Scholar] [CrossRef] [PubMed]
- Kaya, B.; Erdi, F.; Kilinc, I.; Keskin, F.; Feyzioglu, B.; Esen, H.; Karatas, Y.; Uyar, M.; Kalkan, E. Alterations of the thioredoxin system during subarachnoid hemorrhage-induced cerebral vasospasm. Acta Neurochir. 2015, 157, 793–800. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Bharti, V.; Zhou, H.; Hoi, V.; Tan, H.; Wu, Z.; Nagakannan, P.; Eftekharpour, E.; Wang, J.F. Upregulation of Thioredoxin-Interacting Protein in Brain of Amyloid-beta Protein Precursor/Presenilin 1 Transgenic Mice and Amyloid-beta Treated Neuronal Cells. J. Alzheimers Dis. 2019, 72, 139–150. [Google Scholar] [CrossRef]
- Ismael, S.; Wajidunnisa; Sakata, K.; McDonald, M.P.; Liao, F.F.; Ishrat, T. ER stress associated TXNIP-NLRP3 inflammasome activation in hippocampus of human Alzheimer’s disease. Neurochem. Int. 2021, 148, 105104. [Google Scholar] [CrossRef]
- Li, L.; Ismael, S.; Nasoohi, S.; Sakata, K.; Liao, F.F.; McDonald, M.P.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) Associated NLRP3 Inflammasome Activation in Human Alzheimer’s Disease Brain. J. Alzheimers Dis. 2019, 68, 255–265. [Google Scholar] [CrossRef]
- Zhao, Q.; Che, X.; Zhang, H.; Tan, G.; Liu, L.; Jiang, D.; Zhao, J.; Xiang, X.; Sun, X.; He, Z. Thioredoxin-Interacting Protein Mediates Apoptosis in Early Brain Injury after Subarachnoid Haemorrhage. Int. J. Mol. Sci. 2017, 18, 854. [Google Scholar] [CrossRef]
- Park, H.S.; Song, J.W.; Park, J.H.; Lim, B.K.; Moon, O.S.; Son, H.Y.; Lee, J.H.; Gao, B.; Won, Y.S.; Kwon, H.J. TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy 2021, 17, 2549–2564. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Won, Y.S.; Suh, H.W.; Jeon, J.H.; Shao, Y.; Yoon, S.R.; Chung, J.W.; Kim, T.D.; Kim, H.M.; Nam, K.H.; et al. Vitamin D3 upregulated protein 1 suppresses TNF-alpha-induced NF-kappaB activation in hepatocarcinogenesis. J. Immunol. 2010, 185, 3980–3989. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Won, Y.S.; Nam, K.T.; Yoon, Y.D.; Jee, H.; Yoon, W.K.; Nam, K.H.; Kang, J.S.; Han, S.U.; Choi, I.P.; et al. Vitamin D(3) upregulated protein 1 deficiency promotes N-methyl-N-nitrosourea and Helicobacter pylori-induced gastric carcinogenesis in mice. Gut 2012, 61, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Yang, J.W.; Kwon, J.H.; Lee, H.; Yoon, Y.D.; Choi, B.J.; Lee, M.Y.; Lee, C.W.; Han, S.B.; Kang, J.S. Targeted Induction of Endogenous VDUP1 by Small Activating RNA Inhibits the Growth of Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 7743. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lopez-Ramos, D.A.; Yoshihara, E.; Maeda, Y.; Masutani, H.; Sugie, K.; Maeda, M.; Yodoi, J. Thioredoxin-binding protein-2 (TBP-2/VDUP1/TXNIP) regulates T-cell sensitivity to glucocorticoid during HTLV-I-induced transformation. Leukemia 2011, 25, 440–448. [Google Scholar] [CrossRef]
- Jiang, N.; Liu, J.; Guan, C.; Ma, C.; An, J.; Tang, X. Thioredoxin-interacting protein: A new therapeutic target in bone metabolism disorders? Front. Immunol. 2022, 13, 955128. [Google Scholar] [CrossRef]
- Takahashi, Y.; Masuda, H.; Ishii, Y.; Nishida, Y.; Kobayashi, M.; Asai, S. Decreased expression of thioredoxin interacting protein mRNA in inflamed colonic mucosa in patients with ulcerative colitis. Oncol. Rep. 2007, 18, 531–535. [Google Scholar] [CrossRef]
- Birchenough, G.M.; Nystrom, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef]
- Dong, F.; Dong, S.; Liang, Y.; Wang, K.; Qin, Y.; Zhao, X. miR-20b inhibits the senescence of human umbilical vein endothelial cells through regulating the Wnt/beta-catenin pathway via the TXNIP/NLRP3 axis. Int. J. Mol. Med. 2020, 45, 847–857. [Google Scholar] [CrossRef]
- Park, Y.J.; Yoon, S.J.; Suh, H.W.; Kim, D.O.; Park, J.R.; Jung, H.; Kim, T.D.; Yoon, S.R.; Min, J.K.; Na, H.J.; et al. TXNIP deficiency exacerbates endotoxic shock via the induction of excessive nitric oxide synthesis. PLoS Pathog. 2013, 9, e1003646. [Google Scholar] [CrossRef]
- Nishizawa, K.; Nishiyama, H.; Matsui, Y.; Kobayashi, T.; Saito, R.; Kotani, H.; Masutani, H.; Oishi, S.; Toda, Y.; Fujii, N.; et al. Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis 2011, 32, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Zhang, F.; Qu, K.; Liu, C.; Zhang, J. TXNIP: A Double-Edged Sword in Disease and Therapeutic Outlook. Oxid. Med. Cell. Longev. 2022, 2022, 7805115. [Google Scholar] [CrossRef]
- Ahlawat, S.; Kumar, P.; Mohan, H.; Goyal, S.; Sharma, K.K. Inflammatory bowel disease: Tri-directional relationship between microbiota, immune system and intestinal epithelium. Crit. Rev. Microbiol. 2021, 47, 254–273. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef] [PubMed]
- Strugala, V.; Dettmar, P.W.; Pearson, J.P. Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn’s disease. Int. J. Clin. Pract. 2008, 62, 762–769. [Google Scholar] [CrossRef]
- Pullan, R.D.; Thomas, G.A.; Rhodes, M.; Newcombe, R.G.; Williams, G.T.; Allen, A.; Rhodes, J. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 1994, 35, 353–359. [Google Scholar] [CrossRef]
- Allen, A.; Hutton, D.A.; Pearson, J.P. The MUC2 gene product: A human intestinal mucin. Int. J. Biochem. Cell Biol. 1998, 30, 797–801. [Google Scholar] [CrossRef]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Buller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Xiao, Y.T.; Yan, W.H.; Cao, Y.; Yan, J.K.; Cai, W. Neutralization of IL-6 and TNF-alpha ameliorates intestinal permeability in DSS-induced colitis. Cytokine 2016, 83, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Vulliemoz, M.; Brand, S.; Juillerat, P.; Mottet, C.; Ben-Horin, S.; Michetti, P. TNF-Alpha Blockers in Inflammatory Bowel Diseases: Practical Recommendations and a User’s Guide: An Update. Digestion 2020, 101 (Suppl. S1), 16–26. [Google Scholar] [CrossRef] [PubMed]
- Liso, M.; Verna, G.; Cavalcanti, E.; De Santis, S.; Armentano, R.; Tafaro, A.; Lippolis, A.; Campiglia, P.; Gasbarrini, A.; Mastronardi, M.; et al. Interleukin 1beta Blockade Reduces Intestinal Inflammation in a Murine Model of Tumor Necrosis Factor-Independent Ulcerative Colitis. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.D.; Busquets-Cortes, C.; Capo, X.; Tejada, S.; Tur, J.A.; Pons, A.; Sureda, A. Cyclooxygenase-2 Inhibitors as a Therapeutic Target in Inflammatory Diseases. Curr. Med. Chem. 2019, 26, 3225–3241. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, W.S.; Kim, D.O.; Byun, J.E.; Huy, H.; Lee, S.Y.; Song, H.Y.; Park, Y.J.; Kim, T.D.; Yoon, S.R.; et al. Macrophage migration inhibitory factor interacts with thioredoxin-interacting protein and induces NF-kappaB activity. Cell. Signal. 2017, 34, 110–120. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Kostadinova, F.I.; Furuichi, K.; Shamekh, M.M.; Kondo, T.; Wada, T.; Egashira, K.; Mukaida, N. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009, 69, 7884–7892. [Google Scholar] [CrossRef]
- Zuo, B.W.; Yao, W.X.; Fang, M.D.; Ren, J.; Tu, L.L.; Fan, R.J.; Zhang, Y.M. Boris knockout eliminates AOM/DSS-induced in situ colorectal cancer by suppressing DNA damage repair and inflammation. Cancer Sci. 2023, 114, 1972–1985. [Google Scholar] [CrossRef]
- Grill, J.I.; Neumann, J.; Ofner, A.; Marschall, M.K.; Zierahn, H.; Herbst, A.; Wolf, E.; Kolligs, F.T. Dro1/Ccdc80 inactivation promotes AOM/DSS-induced colorectal carcinogenesis and aggravates colitis by DSS in mice. Carcinogenesis 2018, 39, 1176–1184. [Google Scholar] [CrossRef]
- Thapa, P.; Jiang, H.; Ding, N.; Hao, Y.; Alshahrani, A.; Lee, E.Y.; Fujii, J.; Wei, Q. Loss of Peroxiredoxin IV Protects Mice from Azoxymethane/Dextran Sulfate Sodium-Induced Colorectal Cancer Development. Antioxidants 2023, 12, 677. [Google Scholar] [CrossRef]
- Li, B.; Alli, R.; Vogel, P.; Geiger, T.L. IL-10 modulates DSS-induced colitis through a macrophage-ROS-NO axis. Mucosal Immunol. 2014, 7, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Vlantis, K.; Polykratis, A.; Welz, P.S.; van Loo, G.; Pasparakis, M.; Wullaert, A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut 2016, 65, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Seo, E.B.; Lee, S.H.; Cho, C.H.; Kim, S.J.; Kim, S.J.; Kim, H.R.; Ye, S.K. T Cell-Specific Knockout of STAT3 Ameliorates Dextran Sulfate Sodium-Induced Colitis by Reducing the Inflammatory Response. Immune Netw. 2018, 18, e30. [Google Scholar] [CrossRef]
- Sanada, Y.; Mizushima, T.; Kai, Y.; Nishimura, J.; Hagiya, H.; Kurata, H.; Mizuno, H.; Uejima, E.; Ito, T. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS ONE 2011, 6, e23933. [Google Scholar] [CrossRef] [PubMed]
- Horuluoglu, B.H.; Kayraklioglu, N.; Tross, D.; Klinman, D. PAM3 protects against DSS-induced colitis by altering the M2:M1 ratio. Sci. Rep. 2020, 10, 6078. [Google Scholar] [CrossRef]
- Ye, Z.; Zhu, Y.; Tang, N.; Zhao, X.; Jiang, J.; Ma, J.; Zhang, H. alpha7 nicotinic acetylcholine receptor agonist GTS-21 attenuates DSS-induced intestinal colitis by improving intestinal mucosal barrier function. Mol. Med. 2022, 28, 59. [Google Scholar] [CrossRef]
- Lee, K.N.; Kang, H.S.; Jeon, J.H.; Kim, E.M.; Yoon, S.R.; Song, H.; Lyu, C.Y.; Piao, Z.H.; Kim, S.U.; Han, Y.H.; et al. VDUP1 is required for the development of natural killer cells. Immunity 2005, 22, 195–208. [Google Scholar] [CrossRef]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L.; et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef]
- Kang, J.S.; Kim, H.M.; Choi, I.Y.; Han, S.B.; Yoon, Y.D.; Lee, H.; Park, K.H.; Cho, I.J.; Lee, C.W.; Lee, K.; et al. DBM1285 suppresses tumor necrosis factor alpha production by blocking p38 mitogen-activated protein kinase/mitogen-activated protein kinase-activated protein kinase 2 signaling pathway. J. Pharmacol. Exp. Ther. 2010, 334, 657–664. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.H.; Lee, H.; Kim, H.-C.; Choi, I.; Han, S.-B.; Kang, J.S. VDUP1 Deficiency Promotes the Severity of DSS-Induced Colitis in Mice by Inducing Macrophage Infiltration. Int. J. Mol. Sci. 2023, 24, 13584. https://doi.org/10.3390/ijms241713584
Park KH, Lee H, Kim H-C, Choi I, Han S-B, Kang JS. VDUP1 Deficiency Promotes the Severity of DSS-Induced Colitis in Mice by Inducing Macrophage Infiltration. International Journal of Molecular Sciences. 2023; 24(17):13584. https://doi.org/10.3390/ijms241713584
Chicago/Turabian StylePark, Ki Hwan, Hyunju Lee, Hyoung-Chin Kim, Inpyo Choi, Sang-Bae Han, and Jong Soon Kang. 2023. "VDUP1 Deficiency Promotes the Severity of DSS-Induced Colitis in Mice by Inducing Macrophage Infiltration" International Journal of Molecular Sciences 24, no. 17: 13584. https://doi.org/10.3390/ijms241713584