
Activation of G-Protein-Coupled Estrogen Receptor 1 (GPER1) Reduces Progression of Vulvar Carcinoma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
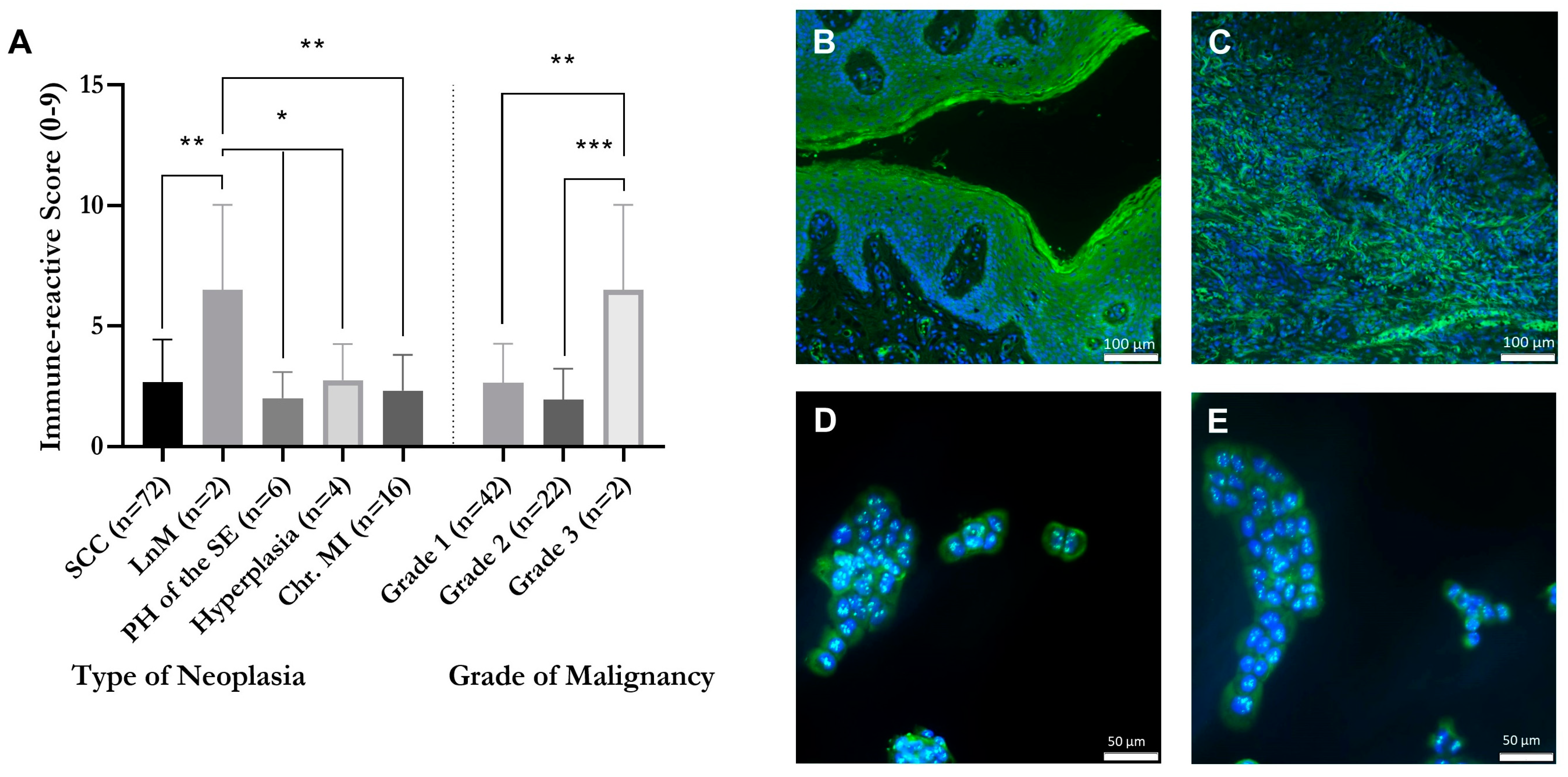
2.1. Correlation of GPER1 Expression and Grade of Malignancy within a Tissue Microarray of Vulvar Neoplasia
2.2. Detection of GPER1 in Vulvar Carcinoma Cells in Cytoplasm and Nucleus
2.3. G1 Acts through GPER1 in Vulvar Carcinoma Cells
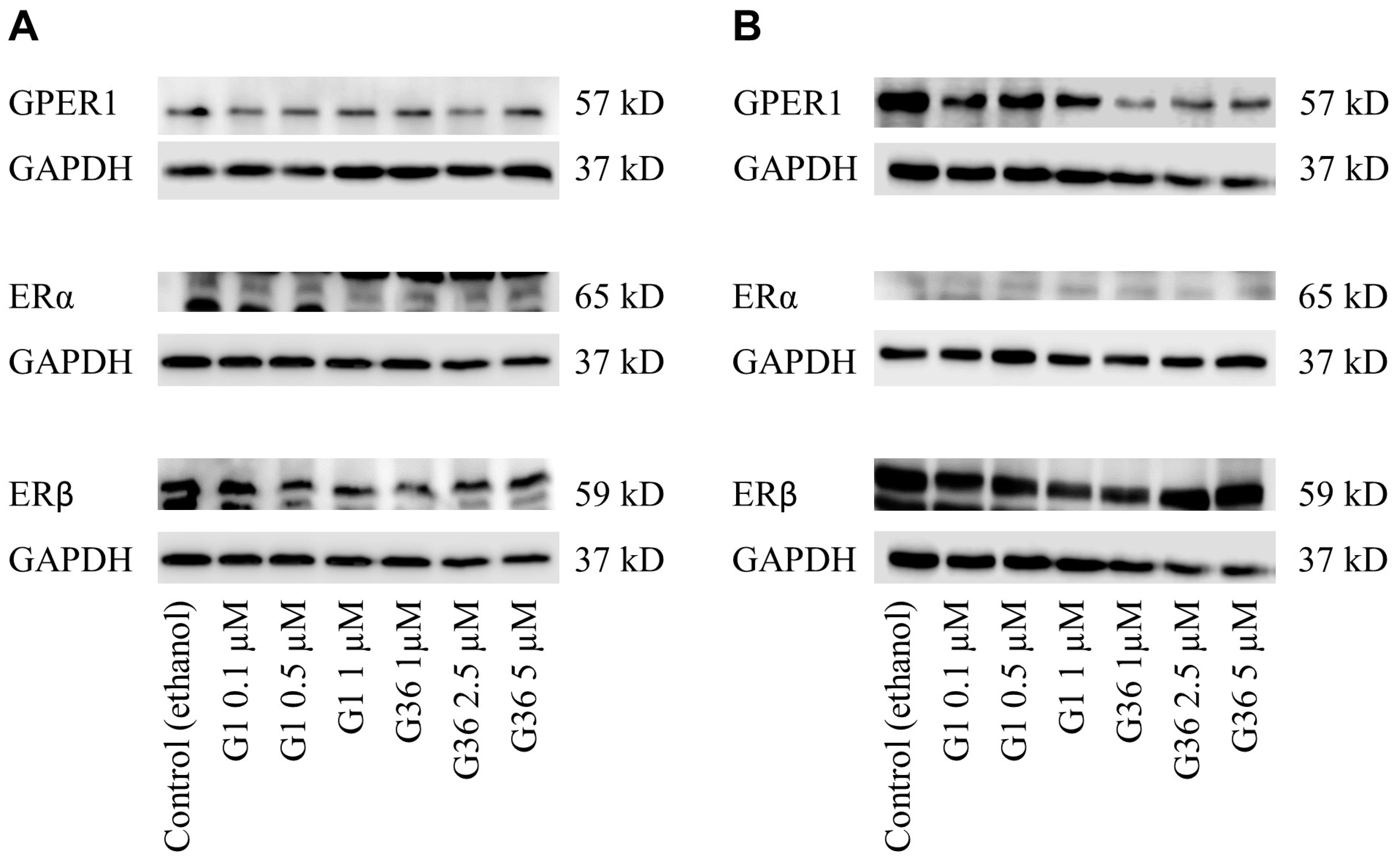
2.4. No Impact of GPER1 Agonist G1 and Antagonist G36 Treatment on Expression of Estrogen Receptors in Vulvar Carcinoma Cells
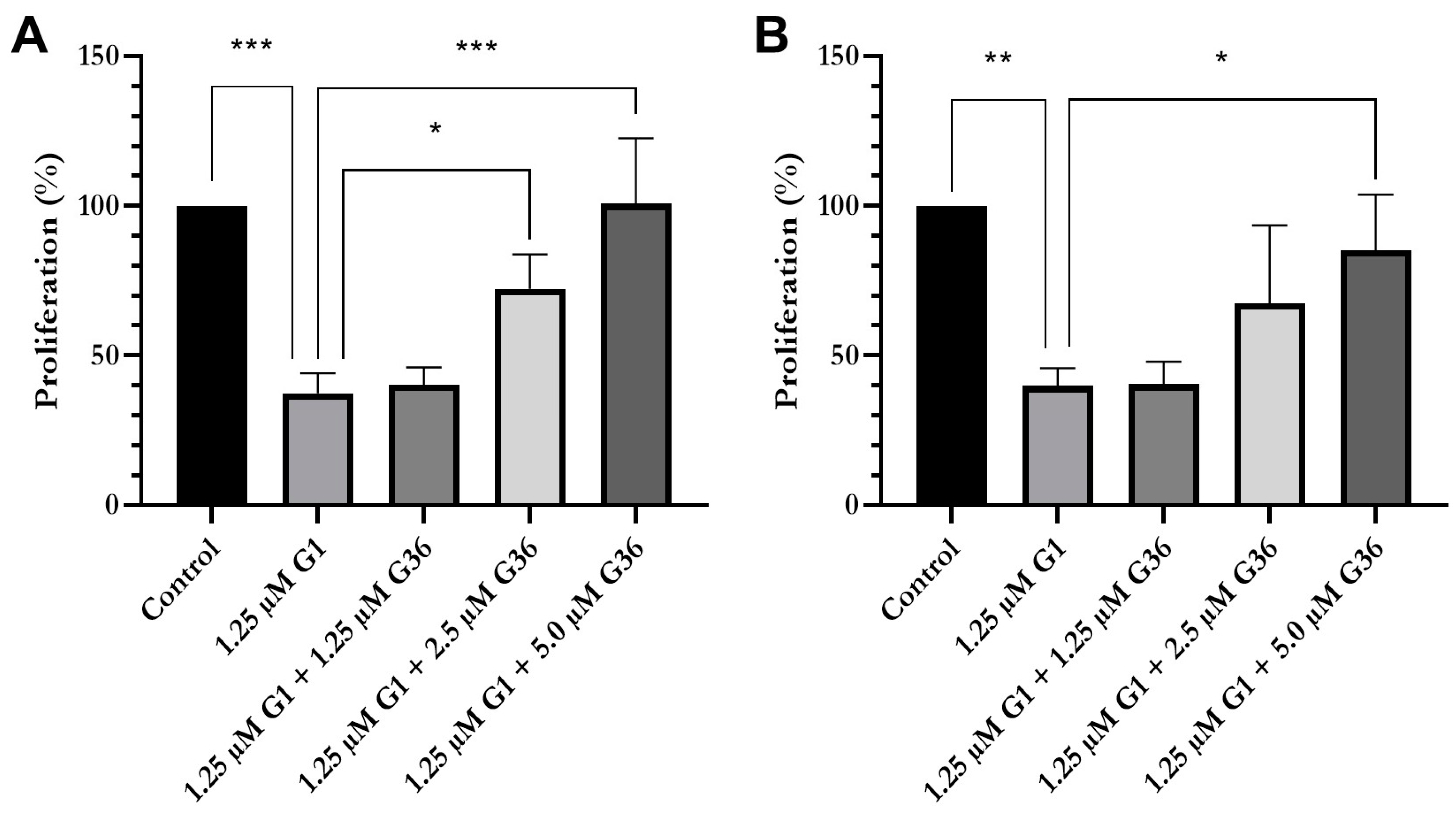
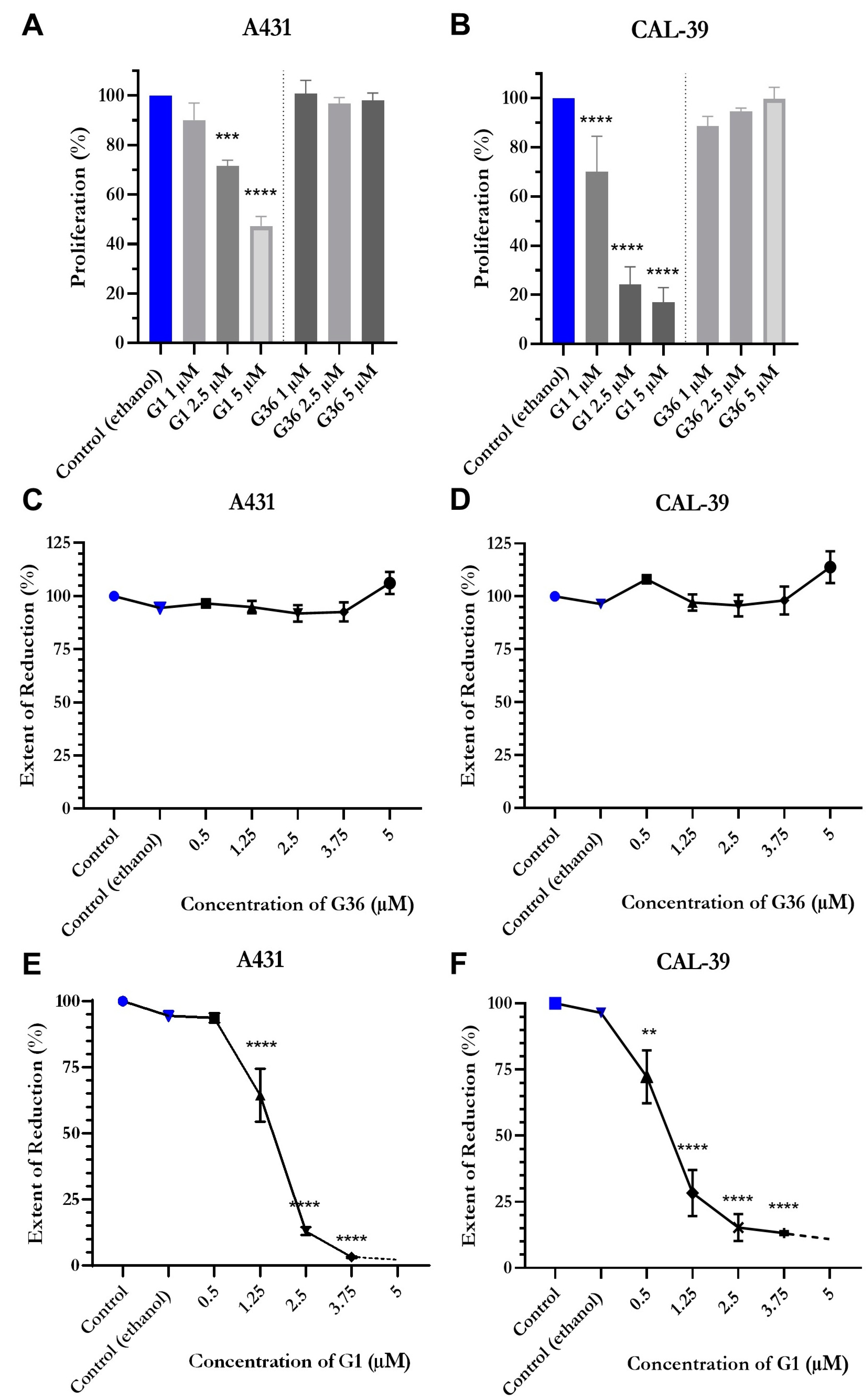
2.5. Reduced Proliferation and Viability after Treatment with GPER1 Agonist G1
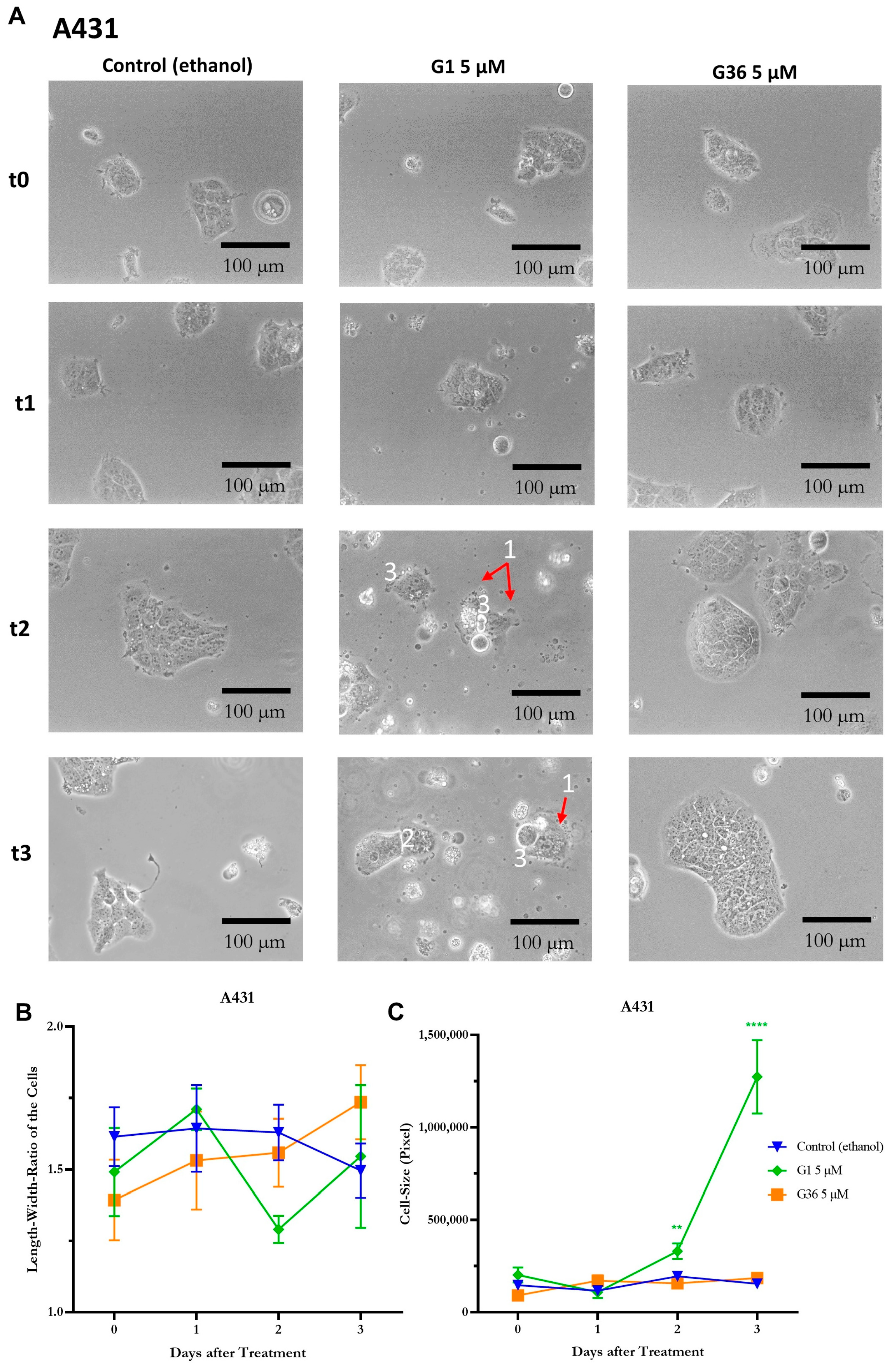
2.6. Signs of Necrosis after Treatment with GPER1 Agonist G1
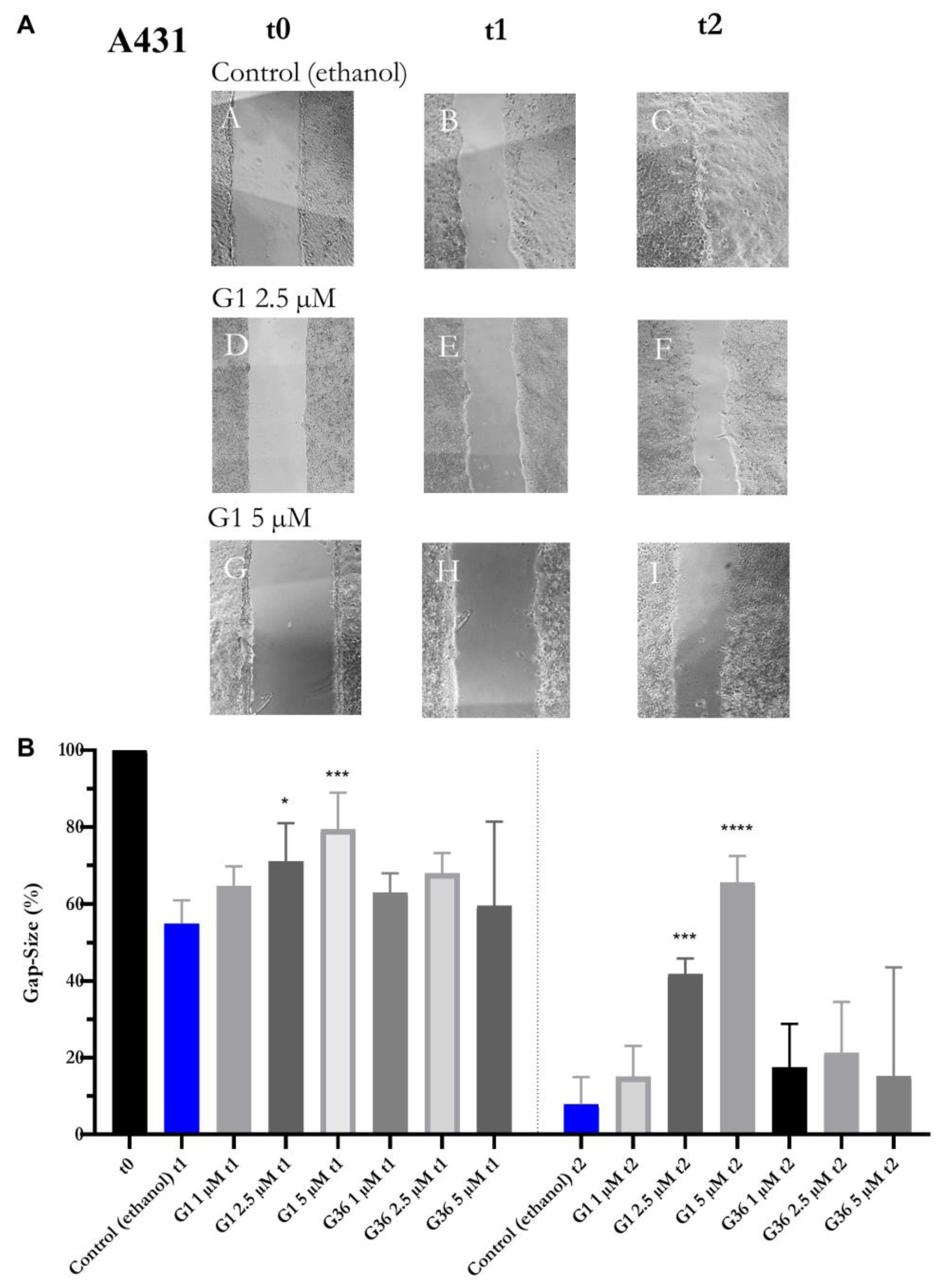
2.7. Effects of GPER1 Agonist G1 and Antagonist G36 on Migration of Vulvar Carcinoma Cells
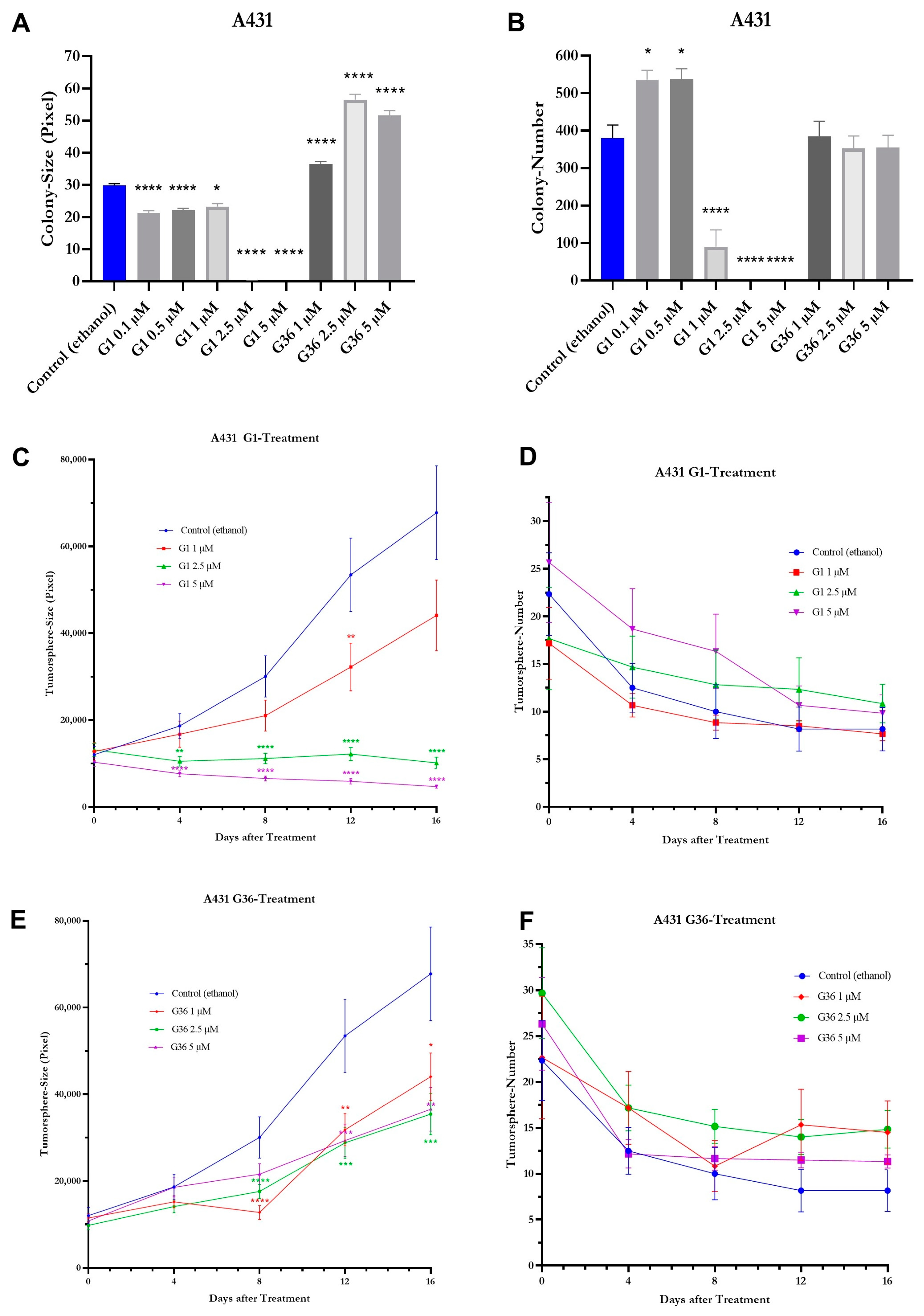
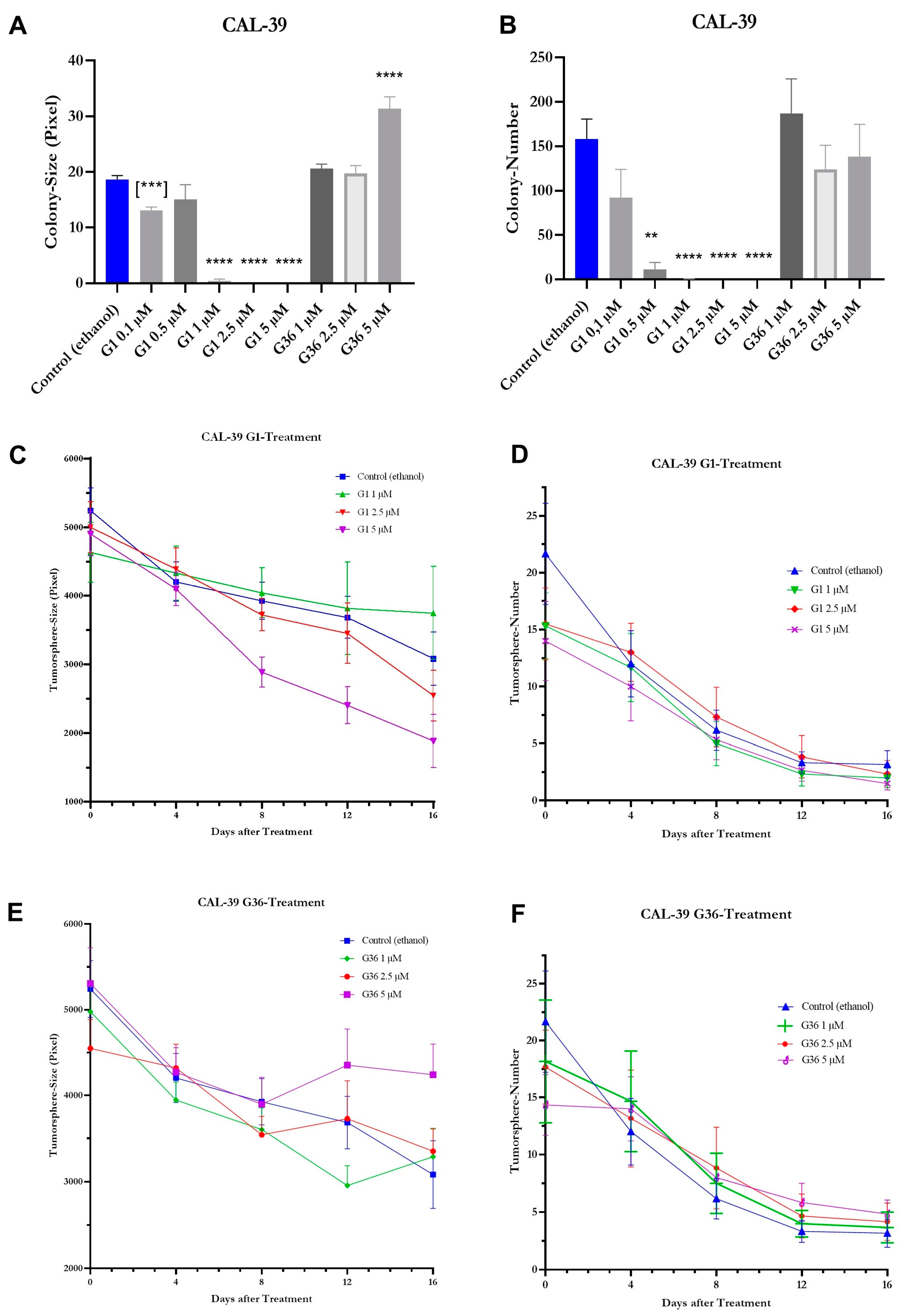
2.8. Effects of GPER1 Agonist G1 and Antagonist G36 on Colony Formation and Sphere Formation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Drugs
4.3. Proliferation
4.4. Viability
4.5. Cell Morphology
4.6. Gap Closure
4.7. Colony Formation
4.8. Tumor Sphere Formation
4.9. Tissue Microarray
4.10. Immune Cytology
4.11. Western Blot
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldnau, C.; Köninger, A.; Kimmig, R. Das äußere Genitale der Frau: Pathologische Befunde und initiale Behandlungsschritte. Der Urol. 2010, 49, 1496–1502. [Google Scholar] [CrossRef]
- Hacker, N.F.; Eifel, P.J.; van der Velden, J. Cancer of the vulva. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 2015, 131 (Suppl. 2), S76–S83. [Google Scholar] [CrossRef]
- Merlo, S. Modern treatment of vulvar cancer. Radiol. Oncol. 2020, 54, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Nugent, E.K.; Brooks, R.A.; Barr, C.D.; Case, A.S.; Mutch, D.G.; Massad, L.S. Clinical and Pathologic Features of Vulvar Intraepithelial Neoplasia in Premenopausal and Postmenopausal Women. J. Low. Genit. Tract Dis. 2011, 15, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Guidozzi, F. Estrogen therapy in gynecological cancer survivors. Climacteric 2013, 16, 611–617. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, J.; van Lindert, A.C.; Gimbrere, C.H.; Oosting, H.; Heintz, A.M. Epidemiologic Data on Vulvar Cancer: Comparison of Hospital with Population-Based Data. Gynecol. Oncol. 1996, 62, 379–383. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, R.; Sant, M.; Coleman, M.P.; Francisci, S.; Baili, P.; Pierannunzio, D.; Trama, A.; Visser, O.; Brenner, H.; Ardanaz, E.; et al. Cancer survival in Europe 1999-2007 by country and age: Results of EUROCARE-5-A population-based study. Lancet Oncol. 2014, 15, 23–34. [Google Scholar] [CrossRef]
- Preti, M.; Bucchi, L.; Micheletti, L.; Privitera, S.; Corazza, M.; Cosma, S.; Gallio, N.; Borghi, A.; Bevilacqua, F.; Benedetto, C. Four-decade trends in lymph node status of patients with vulvar squamous cell carcinoma in northern Italy. Sci. Rep. 2021, 11, 5661. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef]
- Hernandez-Silva, C.D.; Riera-Leal, A.; Ortiz-Lazareno, P.C.; Jave-Suárez, L.F.; Ramírez de Arellano, A.; Lopez-Pulido, E.I.; Macías-Barragan, J.G.; Montoya-Buelna, M.; Dávila-Rodríguez, J.R.; Chabay, P.; et al. GPER Overexpression in Cervical Cancer Versus Premalignant Lesions: Its Activation Induces Different Forms of Cell Death. Anti-Cancer Agents Med. Chem. 2019, 19, 783–791. [Google Scholar] [CrossRef]
- Ino, Y.; Akimoto, T.; Takasawa, A.; Takasawa, K.; Aoyama, T.; Ueda, A.; Ota, M.; Magara, K.; Tagami, Y.; Murata, M.; et al. Elevated expression of G protein-coupled receptor 30 (GPR30) is associated with poor prognosis in patients with uterine cervical adenocarcinoma. Histol. Histopathol. 2020, 35, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Gao, X.-H.; Kaul, R. Estrogen receptor subtype agonist activation in human cutaneous squamous cell carcinoma cells modulates expression of CD55 and Cyclin D1. EXCLI J. 2019, 18, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.-Y.; Weng, J.-R.; Hu, J.-L.; Wang, D.; Sargeant, A.M.; Chiu, C.-F. G15, a GPR30 antagonist, induces apoptosis and autophagy in human oral squamous carcinoma cells. Chem. Interactions 2013, 206, 375–384. [Google Scholar] [CrossRef]
- Hernández-Silva, C.D.; Villegas-Pineda, J.C.; Pereira-Suárez, A.L. Expression and Role of the G Protein-Coupled Estrogen Receptor (GPR30/GPER) in the Development and Immune Response in Female Reproductive Cancers. Front. Endocrinol. 2020, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Jacenik, D.; Cygankiewicz, A.I.; Krajewska, W.M. The G protein-coupled estrogen receptor as a modulator of neoplastic transformation. Mol. Cell. Endocrinol. 2016, 429, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Emons, G.; Gründker, C. Estrogen Signaling in ERα-Negative Breast Cancer: ERβ and GPER. Front. Endocrinol. 2019, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.-I.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef]
- Ku, T.K.S.; Crowe, D.L. Coactivator-mediated estrogen response in human squamous cell carcinoma lines. J. Endocrinol. 2007, 193, 147–155. [Google Scholar] [CrossRef]
- Mancuso, M.; Gallo, D.; Leonardi, S.; Pierdomenico, M.; Pasquali, E.; De Stefano, I.; Rebessi, S.; Tanori, M.; Scambia, G.; Di Majo, V.; et al. Modulation of basal and squamous cell carcinoma by endogenous estrogen in mouse models of skin cancer. Carcinog. 2009, 30, 340–347. [Google Scholar] [CrossRef]
- Han, N.; Heublein, S.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; et al. The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro. Cells 2021, 10, 619. [Google Scholar] [CrossRef]
- Girgert, R.; Emons, G.; Gründker, C. Inhibition of growth hormone receptor by Somavert reduces expression of GPER and prevents growth stimulation of triple-negative breast cancer by 17β-estradiol. Oncol. Lett. 2018, 15, 9559–9566. [Google Scholar] [CrossRef]
- Liu, Y.; Du, F.-Y.; Chen, W.; Fu, P.-F.; Yao, M.-Y.; Zheng, S.-S. G15 sensitizes epithelial breast cancer cells to doxorubicin by preventing epithelial-mesenchymal transition through inhibition of GPR30. Am. J. Transl. Res. 2015, 7, 967–975. [Google Scholar]
- Molina, L.; Bustamante, F.; Ortloff, A.; Ramos, I.; Ehrenfeld, P.; Figueroa, C.D. Continuous Exposure of Breast Cancer Cells to Tamoxifen Upregulates GPER-1 and Increases Cell Proliferation. Front. Endocrinol. 2020, 11, 563165. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, V.; Bauerschmitz, G.; Gallwas, J.; Gründker, C. Suppression of G Protein-coupled Estrogen Receptor 1 (GPER1) Enhances the Anti-invasive Efficacy of Selective ERβ Agonists. Anticancer. Res. 2022, 42, 5187–5194. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-Y.V.; Lee, J.-W.; Kim, W.-S.; Jung, K.-L.; Lee, S.-J.; Lee, J.-H.; Bae, D.-S.; Kim, B.-G. Adenoid cystic carcinoma of the Bartholin's gland: Report of two cases and review of the literature. Gynecol. Oncol. 2006, 100, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Friese, K.; Kost, B.; Vattai, A.; Marmé, F.; Kuhn, C.; Mahner, S.; Dannecker, C.; Jeschke, U.; Heublein, S. The G protein-coupled estrogen receptor (GPER/GPR30) may serve as a prognostic marker in early-stage cervical cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-López, I.G.; De Arellano, A.R.; Jave-Suárez, L.F.; Hernández-Silva, C.D.; García-Chagollan, M.; Hernández-Bello, J.; Lopez-Pulido, E.I.; Macias-Barragan, J.; Montoya-Buelna, M.; Muñoz-Valle, J.F.; et al. Interaction between 17β-estradiol, prolactin and human papillomavirus induce E6/E7 transcript and modulate the expression and localization of hormonal receptors. Cancer Cell Int. 2019, 19, 227. [Google Scholar] [CrossRef] [PubMed]
- Ruckriegl, S.; Loris, J.; Wert, K.; Bauerschmitz, G.; Gallwas, J.; Gründker, C. Knockdown of G Protein-coupled Estrogen Receptor 1 (GPER1) Enhances Tumor-supportive Properties in Cervical Carcinoma Cells. Cancer Genom. Proteom. 2023, 20, 281–297. [Google Scholar] [CrossRef]
- Akimoto, T.; Takasawa, A.; Takasawa, K.; Aoyama, T.; Murata, M.; Osanai, M.; Saito, T.; Sawada, N. Estrogen/GPR30 Signaling Contributes to the Malignant Potentials of ER-Negative Cervical Adenocarcinoma via Regulation of Claudin-1 Expression. Neoplasia 2018, 20, 1083–1093. [Google Scholar] [CrossRef]
- Zhu, C.-X.; Xiong, W.; Wang, M.-L.; Yang, J.; Shi, H.-J.; Chen, H.-Q.; Niu, G. Nuclear G protein-coupled oestrogen receptor (GPR30) predicts poor survival in patients with ovarian cancer. J. Int. Med. Res. 2018, 46, 723–731. [Google Scholar] [CrossRef]
- Gerdes, J. Ki-67 and other proliferation markers useful for immunohistological diagnostic and prognostic evaluations in human malignancies. Semin. Cancer Biol. 1990, 1, 199–206. [Google Scholar]
- Camp, R.L.; Charette, L.A.; Rimm, D.L. Validation of Tissue Microarray Technology in Breast Carcinoma. Lab. Investig. 2000, 80, 1943–1949. [Google Scholar] [CrossRef]
- Lv, X.; He, C.; Huang, C.; Hua, G.; Wang, Z.; Remmenga, S.W.; Rodabough, K.J.; Karpf, A.R.; Dong, J.; Davis, J.S.; et al. G-1 Inhibits Breast Cancer Cell Growth via Targeting Colchicine-Binding Site of Tubulin to Interfere with Microtubule Assembly. Mol. Cancer Ther. 2017, 16, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Ito, F.; Matsushima, H.; Takaoka, O.; Tanaka, Y.; Koshiba, A.; Kusuki, I.; Kitawaki, J. G protein–coupled estrogen receptor 1 agonist G-1 induces cell cycle arrest in the mitotic phase, leading to apoptosis in endometriosis. Fertil. Steril. 2015, 103, 1228–1235.e1221. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lv, X.; Jiang, C.; Davis, J.S. The putative G-protein coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian and breast cancer cells in a GPER-independent manner. Am. J. Transl. Res. 2012, 4, 390–402. [Google Scholar] [PubMed]
- Padet, L.; St-Amour, I.; Aubin, E.; Proulx, D.P.; Bazin, R.; Lemieux, R. Dose-Dependent inhibition of brdu detection in the cell proliferation elisa by culture medium proteins. J. Immunoass. Immunochem. 2009, 30, 348–357. [Google Scholar] [CrossRef]
- Girgert, R.; Emons, G.; Gründker, C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: Possible application in targeted therapy. Breast Cancer Res. Treat. 2012, 134, 199–205. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2016. [Google Scholar]
- Van Horssen, R.; Hagen, T.L.T. Crossing barriers: The new dimension of 2D cell migration assays. J. Cell. Physiol. 2011, 226, 288–290. [Google Scholar] [CrossRef]
- Horibata, S.; Vo, T.V.; Subramanian, V.; Thompson, P.R.; Coonrod, S.A. Utilization of the Soft Agar Colony Formation Assay to Identify Inhibitors of Tumorigenicity in Breast Cancer Cells. J. Vis. Exp. 2015, 99, e52727. [Google Scholar] [CrossRef]
- Lee, C.H.; Yu, C.C.; Wang, B.Y.; Chang, W.W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2015, 7, 1215–1226. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loris, J.; Hanesch, L.; Bauerschmitz, G.; Gallwas, J.; Gründker, C. Activation of G-Protein-Coupled Estrogen Receptor 1 (GPER1) Reduces Progression of Vulvar Carcinoma Cells. Int. J. Mol. Sci. 2023, 24, 13705. https://doi.org/10.3390/ijms241813705
Loris J, Hanesch L, Bauerschmitz G, Gallwas J, Gründker C. Activation of G-Protein-Coupled Estrogen Receptor 1 (GPER1) Reduces Progression of Vulvar Carcinoma Cells. International Journal of Molecular Sciences. 2023; 24(18):13705. https://doi.org/10.3390/ijms241813705
Chicago/Turabian StyleLoris, Johanna, Lena Hanesch, Gerd Bauerschmitz, Julia Gallwas, and Carsten Gründker. 2023. "Activation of G-Protein-Coupled Estrogen Receptor 1 (GPER1) Reduces Progression of Vulvar Carcinoma Cells" International Journal of Molecular Sciences 24, no. 18: 13705. https://doi.org/10.3390/ijms241813705
APA StyleLoris, J., Hanesch, L., Bauerschmitz, G., Gallwas, J., & Gründker, C. (2023). Activation of G-Protein-Coupled Estrogen Receptor 1 (GPER1) Reduces Progression of Vulvar Carcinoma Cells. International Journal of Molecular Sciences, 24(18), 13705. https://doi.org/10.3390/ijms241813705