Abstract
Parkinson’s disease (PD) is one of the large-scale health issues detrimental to human quality of life, and current treatments are only focused on neuroprotection and easing symptoms. This study evaluated in silico binding activity and estimated the stability of major metabolites in the roots of R. palmatum (RP) with main protein targets in Parkinson’s disease and their ADMET properties. The major metabolites of RP were subjected to molecular docking and QSAR with α-synuclein, monoamine oxidase isoform B, catechol o-methyltransferase, and A2A adenosine receptor. From this, emodin had the greatest binding activity with Parkinson’s disease targets. The chemical stability of the selected compounds was estimated using density functional theory analyses. The docked compounds showed good stability for inhibitory action compared to dopamine and levodopa. According to their structure–activity relationship, aloe-emodin, chrysophanol, emodin, and rhein exhibited good inhibitory activity to specific targets. Finally, mediocre pharmacokinetic properties were observed due to unexceptional blood–brain barrier penetration and safety profile. It was revealed that the major metabolites of RP may have good neuroprotective activity as an additional hit for PD drug development. Also, an association between redox-mediating and activities with PD-relevant protein targets was observed, potentially opening discussion on electrochemical mechanisms with biological functions.
1. Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease after Alzheimer’s disease [1,2]. It has been considered the world’s fastest-growing neurological disorder and an economic burden for older adults because of medication costs and productivity losses [3]. This disease is characterized by bradykinesia, resting tremors, muscle stiffness, speech and writing changes, postural instability, and a wide array of motor and non-motor symptoms [4,5,6]. While the exact pathogenesis of PD is still unclear, primary observations reveal that such progressive disease develops from the degradation of dopaminergic neurons in the substantia nigra pars compacta (SNpc) in the basal ganglia, and the development of intraneuronal proteinaceous cytoplasmic inclusions called Lewy bodies [7]. Approximately four of five PD patients are idiopathic; the others are presumed of genetic origins [4]. Unfortunately, current treatments for PD are for neuroprotection and symptomatic therapy due to a vague understanding of its exact pathogenesis [5,8].
Dopamine (a catecholamine) is a crucial target for treating symptoms of PD since it is the primary neurotransmitter for movement coordination and control (in the motor circuitry of the brain in the basal ganglia), as well as in the cognition and reward system [9]. The death of dopaminergic neurons in SNpc consequently decreases dopamine production responsible for the motor symptoms of PD, such as its effect on the degradation of the nigrostriatal pathway responsible for sensory stimuli and movement, causing bradykinesia [9,10]. Because of the altered motor circuitry in the basal ganglia, automatic postural adjustments are disrupted, causing postural instability and immobility [11]. Also, thalamic activity from basal ganglia dysfunction was found to be promoted due to decreased dopamine, which regulates the thalamocortical loop, and led to abnormal cortical oscillations that cause resting tremors [12,13]. While the death of dopaminergic neurons is vital in PD indications, no complete conclusions on neuronal death have been reached. However, prior studies hypothesized that neuroinflammation from oxidative stresses, metabolic alterations, mitochondrial dysfunction, the impairment of autophagy functions, and intestinal inflammation from gut dysbiosis affecting the enteric nervous system (ENS) are some of the fundamental mechanisms of neuronal death [5,14,15,16].
Some specified mechanisms of neuronal death are the loss of mitochondrial complex I functionality responsible for dopamine production and fragmented mitochondria from mutations of coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2) [17,18,19]. Also, dysregulations in the tryptophan (Trp)–kynurenine (KYN) metabolic system observed in PD patients potentially led to further neuroinflammation due to the immunomodulatory properties of the metabolites, as well as the accumulation of neuronal death-causing compounds [17,20]. Indeed, imbalances in the Trp-KYN pathway were suspected of causing neurodegeneration due to a heightened ratio of neurotoxic to neuroprotective metabolites, such as the reported increase in quinolinic acid (QUIN) in blood plasma and 3-hydroxykynurenine (3-HK) in the cerebrospinal fluid of PD patients, which was implicated to increased oxidative stress, excitotoxicity, and neuroinflammation where dopaminergic neurons were particularly vulnerable [17,21,22]. Since dopamine is a precursor of other catecholamines, specifically epinephrine and norepinephrine, other non-motor functions—such as peristalsis, heart rate, and blood pressure regulation—are diminished for PD patients [10,23,24,25].
Gnanaraj et al. [8] summarized the PD-relevant proteins of utmost concern for their symptomatic treatment and neuroprotection. One of PD’s first genetic links is the overexpression of SNCA, an α-synuclein (ASN) coding gene, causing the formation of Lewy bodies from ASN aggregations [26]. On the other hand, in the standard treatment of PD, monoamine oxidase isoform B (MAOB) and catechol o-methyltransferase (COMT) are most considered for their function in dopamine metabolism [27]. Due to the impact of dopamine on human movement and control, its excessive decrease promotes uncontrolled and nonrigid movements. At the same time, the antagonism of the A2A adenosine receptor (A2AAR) reduces the wear-off effect of dopaminergic therapies [28]. This receptor modulates motor circuits through inhibition of the direct pathway (motion-induction) and promotion of the indirect pathway (motion-arrest) [28,29]. Current treatments of PD include, but are not limited to, levodopa and its combination drug (such as carbidopa); dopamine agonists (such as apomorphine, ropinirole, cabergoline); MAOB inhibitors (such as rasagiline and selegiline); COMT inhibitors (such as entacapone and tolcapone); anticholinergics; anti-inflammatory agents; and vitamins A, E, and C [5,30].
Natural products have always been an abundant and diverse source of hit and lead compounds with good pharmacokinetic properties, including for treating neurological diseases [31,32]. Interestingly, complex mixtures of these medicines could provide curative potential to PD because of its multiple targeted approach from several potentially bioactive compounds. In traditional Chinese medicine, the roots of Rheum palmatum (RP) or Chinese rhubarb are used for various therapeutic uses, including PD symptom treatment [33]. Previously, it has been revealed that anthraquinones (specifically chrysophanol, rhein, aloe-emodin, emodin, and physcion as shown in Figure 1) are abundant in RP [34]. Consequently, neurotransmitters such as dopamine, epinephrine, norepinephrine, and levodopa show redox-mediating activity for microbial fuel cells (MFCs) and quasi-reversible redox ability in cyclic voltammetry measurements [35]. Also, PD medications were observed as quasi-reversible redox mediators where the electrochemical mechanism for biological function is still open for follow-up investigations [36]. Since RP extracts show good electron-shuttling characteristics, the major metabolites of RP may have similar activity as the treatment mentioned [34]. Indeed, prior studies have explored that emodin and chrysophanol exhibited neuroprotective properties in vitro and in vivo for PD [37].
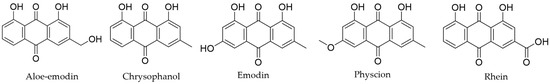
Figure 1.
Chemical structures of RP major metabolites.
A molecular simulation study was conducted for RP’s major metabolites, revealing the potential alternative therapeutic compounds or conjunctive use of RP extracts with conventional medicines for PD. In silico investigations of major compounds of RP were performed, considering neurotransmitters, and PD medicines have good MFC bioenergy amplification. This investigation evaluated their potencies to relevant PD targets and safety profiles with their binding affinity, pharmacokinetic properties, expected reactivity, and quantitative structure–activity relationship for the prediction of their inhibitory action.
2. Results and Discussion
2.1. Molecular Docking
Primary metabolites from RP were subjected to the ligand preparation protocol in Discovery Studio, which generated the 31 ligands presented in Table S1. Two standard drugs used for PD treatment (dopamine and levodopa) were used as positive controls and were prepared similarly to the mentioned metabolites. Validation of the protein structures of protein targets for PD revealed that the root-mean-square-difference (RMSD) of minimized structures compared to experimentally elucidated structures were no more than 2.0 Å, which indicated valid structures for docking studies (see Table S2) [38,39]. Each ligand form was screened using hotspots-based docking, using the LibDock protocol available in Discovery Studio reported in Table 1. The ligand conformers for each metabolite with the highest LibDock score were selected for further analysis to CDOCKER, a CHARMm-based docking protocol.

Table 1.
LibDock scores of RP major metabolites to PD targets.
Furthermore, 35 protein–ligand complexes were refined through CDOCKER with the top hits presented in Table 2, where CDOCKER energy combined protein–ligand binding interaction energy and ligand conformational energy. Compounds above or within the dopamine and levodopa standard range were analyzed for their molecular interaction and properties. While binding these metabolites rendered them inferior to levodopa, multiple ligands inhibited many targets. In this condition, emodin exhibited the best binding activity among all protein targets. Specifically, ASN (PDB ID: 1XQ8) interacted the best with emodin (−34.4961 kcal/mol), surpassing dopamine (−31.0394 kcal/mol). There are only functional assumptions of ASN on dopamine regulation from its ability for synaptic vessel provision in presynaptic terminals and cytoskeletal dynamics from microtubular interaction [40,41]. Interestingly, one symptomatic progression of PD concerns alternate folding of ASN forming insoluble fibrils, which further aggregates leading to neurodegeneration, and they are postulated as causative to prion-like cell-to-cell transmission of ASN misfolding [42]. Nevertheless, the aggregation of ASN is widespread in numerous brain regions of the central nervous system (CNS) for PD patients. These aggregations impair numerous neural functions, such as proteasomal and lysosomal functions, potentially hindering protein clearance and promoting neuronal death [26,43]. Additionally, there are preliminary investigations on ASN aggregation due to CHCHD2 mutations and interaction with 3-HK. Hence, it was suspected that interactions with ASN could inhibit the formation of Lewy bodies for decreased neurotoxicity.
Table 2.
CDOCKER energy (in kcal/mol) of RP major metabolites to PD targets.
Nevertheless, chrysophanol (−39.7554 kcal/mol) and physcion (−30.3292 kcal/mol) may have a good interaction with MAOB (PDB ID: 2C65). At the same time, the best-interacting compound for COMT (PDB ID: 3BWM) is emodin (−32.431 kcal/mol). Normally, MAOB catalyzes the oxidative deamination of xenobiotic and biogenic amines, including dopamine [44]. In contrast, COMT normally regulates levels of catecholamines in the brain and peripheral tissues, especially levodopa, by its inactivation through methyl group transfer from the S-adenosylmethionine to the catechol structure [27,44]. For this reason, their inhibition ameliorates the motor dysfunction and fluctuations observed in PD patients and offsets the deterioration of dopaminergic neurons. Also, emodin has superior binding activity (−57.5427 kcal/mol) to A2AAR (PDB ID: 3EML), comparable to levodopa (−61.8193 kcal/mol). At the same time, rhein (−39.4557 kcal/mol) has a similarity in binding affinity with dopamine (−39.3845 kcal/mol) to A2AAR (PDB ID: 3EML). Due to the inhibitory interaction of A2AAR with the dopamine D2 receptor, reduced availability of dopamine in specific periods exacerbates motor fluctuations [28,29]. Hence, inhibition of A2AAR lessens the worsening of PD symptoms, such as dyskinesia, making it a non-dopaminergic treatment, usually in combination with dopaminergic treatments [28].
Frequent drug–receptor interactions of known drugs, such as hydrophobic contact, hydrogen bonding (HB), and π-stacked interactions [45], were observed in the selected protein–ligand complexes shown in Figure 2. The ASN-emodin complex formed a salt bridge between LYS45 and the alkoxide of the o-dihydroxy aromatic moiety in emodin. Alkyl and π-alkyl interactions were also a contributor to the stability of this complex. For the MAOB-chrysophanol complex, an attractive charge between LYS296 and the alkoxide of the phenolic structure, as well as HB with SER59 and TYR60, caused stabilization of the formed complex. This complex is further stabilized with stacked π-π, π-alkyl, and alkyl interactions. The superior binding of the MAOB-emodin complex formed an attractive charge between LYS296 and the alkoxide and is further stabilized with π-sulfur, stacked π-π, π-alkyl, and alkyl interactions. At the same time, the MAOB-physcion complex formed HB between CYS172 and TYR435 with π-sulfur, stacked π-π, T-shaped π-π, π-alkyl, and alkyl interactions, suggesting excellent molecular orbital alignment. The COMT-emodin complex formed HB between ASP141 and ASN170 and an attractive charge further supported by an attractive charge, π-cation, π-sulfur, π-alkyl, and alkyl interactions. For the A2AAR-emodin complex, salt bridges and attractive charges (ARG107, ARG206, LYS1060, ARG1008), as well as HB (ARG107, ARG199, ARG206, and GLU1005), were formed; however, there was an unfavorable donor–donor interaction (ARG1008) which could potentially destabilize the complex structure. Finally, the A2AAR-rhein complex formed HB (ARG199, LEU202, ALA203, ARG206, and ARG1008), attractive charge (ARG107), and π-alkyl (LEU202 and ALA203). These interactions reveal considerable effects on the PD targets; however, this static simulation could not fully uncover the protein conformation changes that could affect protein–ligand interactions. Hence, further analysis with molecular dynamics simulations could uncover these details.
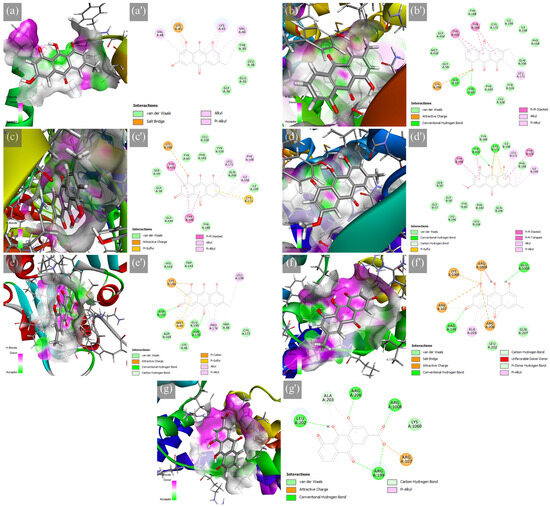
Figure 2.
Hydrogen bond surface of protein receptor and 2D molecular interactions of major RP metabolites to PD targets, which are the (a,a′) ASN-emodin, (b,b′) MAOB-chrysophanol, (c,c′) MAOB-emodin, (d,d′) MAOB-physcion, (e,e′) COMT-emodin, (f,f′) A2AAR-emodin, and (g,g′) A2AAR-rhein.
2.2. Density Functional Theory (DFT) Analysis
The energies of frontier molecular orbitals (FMO) can provide helpful quantum mechanical calculations describing compounds’ chemical reactivity and stability. According to Koopmans’ theorem, the energy of the highest occupied molecular orbital (εHOMO) can be approximated to the required first ionization energy, and the energy of the lowest unoccupied molecular orbital (εLUMO) approximates the electron affinity in the last occupied orbital. In contrast, the energy gap (Δε) relates to the reactivity of the compound [46,47,48,49]. Global reactivity descriptors from approximated ionization energies and electron affinity revealed beneficial molecular properties. In a chemical process, the hardness,
of molecules describes the resistance of electron cloud deformation to trivial perturbations. In contrast, the chemical potential,
shows the stability of the compounds through their electron transfer capability, where negative values indicate the compound for less propensity for decomposition [46,49,50]. The electrophilic index,
is one of the significant descriptors, since it describes potential affinity to a receptor for drug–receptor interactions. This descriptor assesses the inclination of the compound to acquire additional charge and its resistance to electronic charge exchange with the surroundings, indicative of electrophilicity [50]. A large energy band gap between the HOMO and LUMO is classified as hard molecules and is consequently less reactive. Conversely, soft molecules are more reactive for smaller energy band gaps [50,51]. From this, excellent inhibitors are characterized by their heightened stability.
Compared to the minimized structures (in Table S7), all protein–ligand complexes except rhein in complex with A2AAR (PDB ID: 3EML) exhibited an increased energy band gap, revealing increased reactivity. Emodin (2.4536 eV) with ASN (PDB ID: 1XQ8) increased from 2.2949 eV. At the same time, chrysophanol (2.6025 eV), emodin (2.3333 eV), and physcion (3.0351 eV) with MAOB (PDB ID: 2C65) increased from 2.5902, 2.1354, and 2.9551 eV. At the same time, emodin (2.3313 eV and 2.3155 eV) with COMT (PDB ID: 3BWM) and A2AAR (PDB ID: 3EML) increased from 2.1354 eV. However, rhein (2.7222 eV) with A2AAR (PDB ID: 3EML) indicated a decrease from 2.8997 eV. Compared to the standards, MAOB-physcion and A2AAR-rhein exhibited better protein–ligand interaction than levodopa. Similarly, ASN-emodin and MAOB-chrysophanol have potentially more stable protein–ligand interactions. The chemical hardness of the ligands shown in Table 3 indicated less polarizability, where the mentioned are again superior in stability. Moreover, chemical potentials reveal chemical stability from decomposition and electron escaping tendency. All ligands were considered to have superior stability compared to the standards. The electrophilicity indices show rhein and physcion as least nucleophilic.
Table 3.
Electronic properties of standards and docked conformations of ligands (in eV).
2.3. Predicted Inhibitory Activity from 3D QSAR Models
Quantitative structure–activity relationship models were established in this study for the prediction of the inhibitory activity of RP major metabolites on PD targets. Steric (from van der Waals potential of carbon atom probe) and electrostatic (from H+ point charge probe) contributions were considered in the 3D grid space of aligned and minimized ligands with known half-maximal inhibitory concentrations. A dataset of 45 known ASN inhibitors, 140 known MAOB inhibitors, 105 known COMT inhibitors, and 120 known A2AAR inhibitors was considered in the 3D QSAR model building. As shown in Table 4, all models are considered robust and reliable from set significance based on their internal validity. For a further assessment of their accuracy, cross-validation of the models reveals the most accuracy from MAOB (0.488) > A2AAR (0.400) > COMT (0.303) > ASN (0.225). On the other hand, external validation reveals the most accuracy from MAOB (0.458) > COMT (0.433) > ASN (0.387) > A2AAR (0.175). Figures S1–S4 indicate predicted and experimental pIC50 values in the training and test datasets, including their residuals.
Table 4.
3D QSAR model parameters.
All predicted values of IC50 to each protein target are within the dataset range as shown in Table 5. Consistently, emodin (15.957 µM) and rhein (18.457 µM) had the best inhibition of ASN among all metabolites. Compared to epigallocatechin-3-gallate with fibrillogenic inhibitory activity (9.8 µM), the best RP metabolites had 1.63 and 1.88-fold less inhibitory activity [52]. This inhibitory activity on ASN could decrease fibril formation and aggregation, thereby decreasing the toxicity of the oligomeric Lewy bodies to the neurons. Similarly, chrysophanol (0.088 µM) and emodin (0.128 µM) had similar outcomes as their binding affinity with MAOB, which are comparable with selegiline (0.0013 µM), a known MAOB inhibitor [53]. For this, the inhibition of MAOB slows down dopamine degradation, which restores the motor functions of PD patients. Compared to entacapone (0.386 µM) for COMT inhibition [54] and istradefylline (5.25 µM) for A2AAR antagonism [55], the comparable inhibition from aloe-emodin (0.330 µM) and good inhibition with A2AAR of 1.503 µM was revealed, based on its structural features. This interaction could potentially decrease the metabolic activity of levodopa for its increased therapeutic effect and sustained dopamine levels. Also, the decreased blocking activity of A2A receptors to dopamine D2 receptors could potentially restore the balance of the motor circuitry. Nevertheless, chrysophanol (0.816 µM) was shown to have good inhibitory activity with A2AAR. Previous literature experimentally measured the inhibitory concentrations of the major metabolite on MAOB, where only emodin was observed to have appreciable inhibitory activity [56]. However, these results are open to further validations, specifically from immunochemical assays, protein–ligand binding assays, mutagenesis studies, functional assays, and X-ray crystallographic analyses.
Table 5.
Predicted IC50 (in µM) of standards and RP metabolites on PD targets.
2.4. ADMET Properties
Predicted pharmacokinetics and toxicity profiles from structure–activity relationships were performed in ADMETboost and pkCSM, as summarized in Table 6. All major metabolites of RP appeared to satisfy Lipinski’s rule of five. Only chrysophanol did not follow the Pfizer rule, which could potentially have a toxic profile. At the same time, both chrysophanol and physcion do not follow the GSK rule, which may provide subpar ADMET properties. Nevertheless, the physicochemical properties of these metabolites follow drug-like soft rules related to their ease of metabolism without substantially producing bioactive metabolites or intermediates [57]. For this reason, undesired or toxic side effects from metabolic products and drug–drug interactions are minimized. All compounds have desirable n-octanol/water partition coefficients; however, the metabolites are significantly hydrophobic compared to the standards.
Table 6.
ADMET properties in silico with ADMETboost and pkCSM webserver.
Chrysophanol and physcion had good Caco-2 permeability with more than −5.15 log cm/s compared to the standards. On the other hand, all major RP metabolites, including the standards, have excellent human intestinal absorption (with more than 70% probable), which indicates a decreased requirement for parenteral administration. Since traditional medicines commonly use the oral route of administration and aqueous solvent, the good n-octanol/water distribution (1–3 log mol/L) observed for all metabolites and standards and poor aqueous solubility (−4–0.5 log mol/L) could potentially lead to decreased potency of RP but with optimum lipophilicity preferred for medicines for optimal lipid bilayer penetration [59,60]. Nevertheless, the oral bioavailability of the metabolites and standards are comparable with all compounds possessing a good plasma protein binding rate, since they have less than a 90% probability of combining completely with blood serum proteins. This result means that major RP metabolites could potentially have quick onset action, and they can reach the target site rapidly [61,62]. Chrysophanol, physcion, and rhein may cross the blood–brain barrier lightly (between 30–70%) compared to other metabolites and better than levodopa, potentially not requiring advanced drug delivery techniques for therapeutic effect. For these reasons, the metabolites may have good absorption and distribution to affect the required targets appreciably.
The compounds were found to have mediocre CYP2C9 and CYP2D6 inhibitory activities. However, all compounds were revealed to have an excellent probability of CYP2D6 inhibition. The CYP2D6 enzyme has mediocre metabolic activity in these compounds. This enzyme is highly expressed in the SN region of the CNS and is discouraged as a main metabolic route for CNS drugs due to their polymorphic nature [63]. While the half-life of the metabolites was superior to the standards, corresponding to a lengthier action, the hepatocyte clearance was faster than the standards, and the microsome clearance was slower. For this, a cumulative effect may be observed, potentially leading to accumulative ability, and their lipophilicity could have a toxic effect. At the same time, the metabolites reveal a toxicity profile akin to the standards. There is an observed small probability for hERG blocking potential, which could lead to cardiotoxicity. However, this probability could be assumed negligible since the RP metabolites were approximately like the standards. Further investigations into the interaction of the metabolites to hERG with immunochemical assays, molecular simulations, and electrophysiological studies could validate the predicted cardiotoxicity profile. However, mediocre mutagenicity and hepatotoxicity were observed for all metabolites. On the other hand, aloe-emodin, chrysophanol, and physcion were observed to have significantly higher tolerable doses than the other metabolites and the standards, and all metabolites had significantly low lethal doses for oral application. In fact, in vitro and in vivo investigations showed that excessive use of emodin manifested hepatotoxicity, renal toxicity, and reproductive toxicity with chrysophanol [64]. For this reason, the dosage of these compounds requires careful consideration since accumulation could be lethal.
All in silico pharmacokinetics and toxicity property results were consistent with other tools like pkCSM and ADMETlab 2.0. These findings indicate that RP metabolites may have a favorable therapeutic effect as a neuroprotective agent.
3. Materials and Methods
3.1. Preparation of Ligands
This study employed the molecular modeling and visualization software BIOVIA Discovery Studio 2022 (Dassault Systèmes, Vélizy-Villacoublay, France) for molecular simulations. Two-dimensional (2D) structure-data files (SDFs) of the identified ligand molecules were collected from the PubChem database (chem.ncbi.nlm.nih.gov, accessed on 10 May 2023) and prepared with the Prepare Ligands protocol in Discovery Studio (see Table S1). Numerous ionization states were generated under pH 7.5 ± 1.0. A structure visualization of the ligands was generated from ChemDraw v.19.
3.2. Preparation of Proteins
Experimentally elucidated structures of protein targets available from the Protein Data Bank (PDB, www.rcsb.org, accessed on 10 May 2023) in May 2023 were collected in PDB file format. Afterwards, water molecules and irrelevant heteroatoms for the simulation were removed, and polar hydrogens were added to the protein structure. Minimization of the protein structure with co-crystal ligands with the CHARMm forcefield was performed to verify the accuracy of the results. The protein was prepared with the Prepare Protein protocol in Discovery Studio, which automatically repaired and protonated the protein structure under a pH of 7.4 and 0.145 M of ionic strength with 0.9 kcal/mol cutoff energy. Missing loops were built in the protocol using PDB SEQRES loop definition, limited to 20 residues, with loop refinement through CHARMm forcefield minimization.
3.3. Molecular Docking
The initial docking runs were performed using the LibDock algorithm in Discovery Studio to screen ligand forms. Numerous conformations were generated with conjugate-gradient and quasi-newton minimization with an energy threshold between isomers set to 20.0 kcal/mol. A maximum fraction of steric clashes of 10% and a final clustering radius of 0.5 Å were set for the docking. The fractions of acceptable solvent accessible surface area (SASA) cutoffs were set to 15% for apolar SASA and 5% for polar SASA with 18 grid points. The binding sites were determined from the literature and PDB site records (see Table S2) [8,65,66,67,68,69]. Docking optimization was performed using a grid-based CDOCKER algorithm in Discovery Studio for the evaluation of the best ligand for each protein target. Random conformations were generated from 1000 dynamics steps at 1000 K target temperature or with consideration of electrostatic interactions. The docked conformations were refined with 800 maximum bad orientations and a vdW energy threshold of 300 kcal/mol. This complex was annealed for 2000 heating steps until 700 K and subsequently cooled for 5000 steps to 300 K. Final minimizations of the refined ligand pose were set to full potential. The docking analysis results were further evaluated through a comparison to standard drugs of Parkinson’s disease, i.e., dopamine and levodopa [5,30].
3.4. 3D QSAR Modeling and Activity Prediction
The QSAR model for each protein target was modeled after IC50 values of known interacting compounds collected in the ChEMBL database (ebi.ac.uk/chembl, accessed on 10 May 2023) and enumerated in Tables S3–S6. These compounds were prepared using the Prepare Ligands for QSAR protocol in Discovery Studio, where duplicate data were removed, compound structure and charge were repaired, and numerous ionization states were generated under pH 7.4. The compounds were subjected to CHARMm minimization with Momany–Rone ligand partial charge estimation until no energy gradient was reached. Outlier compounds were removed through software-predefined parameters (atomic logarithmic 1-octanol/water partition coefficient, molecular weight, number of H donors, H acceptors, rotatable bonds, rings, aromatic rings, and fractional polar surface area). Outliers were classified under Poisson distribution considering short-ranged parameters with a p-value of 0.01. Rigid molecular overlay for compound alignment was performed under consensus-based, 50% steric, and 50% electrostatic field fit. The IC50 values of the compound data were converted into pIC50 values. These data are separated by diverse split through the predefined parameters into 80% training set and others for the test set. Three-dimensional QSAR models based on shape (steric) and charge (electrostatic) complementarity were generated for each protein target with pIC50 values as the activity variable, grid spacing of 1.5 Å, and 5-fold cross-validation. The 3D QSAR model correlates the relationship between the activity variable and molecular fields following Equation (4):
Variables in this model are n, the number of descriptors which are each grid point; k, the model coefficient for the descriptor; and, V, the descriptor value at the grid point corresponding to the compound. These models are built with partial least squares (PLS) regression.
3.5. Density Functional Theory (DFT) Analysis
Molecular properties of ligand structures in the form of frontier molecular orbitals, band gap (Δε), and electrostatic potential maps were evaluated with the DMol3 package in Discovery Studio for the structure stability analysis. Reference ligands were optimized with CHARMm forcefield minimization. Single-point energy calculation of the reference and docked ligands was performed with Becke’s three-parameter, Lee–Yang–Parr (B3LYP) exchange-correlation functional and aqueous environment using the COSMO continuum solvent models. A double-numeric quality basis set with polarization functions (DNP) and a self-consistent field (SCF) density convergence of 1.0 × 10−6 was used for the analysis.
3.6. ADMET Properties Prediction
SMILES notation records of the prepared ligands were collected from the PubChem database (chem.ncbi.nlm.nih.gov, accessed on 10 May 2023). ADMETboost (ai-druglab.smu.edu/admet, accessed on 10 May 2023) was used for ADMET properties prediction for further analysis. This predictive model utilized MACCS, extended connectivity circular, Mol2Vec, and PubChem fingerprints, as well as Mordred and RDKit descriptors through the extreme gradient boosting machine learning modeling model [58]. Parameters include blood–brain barrier (BBB) permeability, metabolism (inhibition and substrate activity on human cytochrome P450 family), half-life, human ether-a-go-go-related gene (hERG) for cardiotoxicity, human hepatotoxicity, and AMES test for mutagenicity. The human maximum tolerable dose was determined from pkCSM (biosig.lab.uq.edu.au/pkcsm, accessed on 10 May 2023) [70]. Data visualization was generated from GraphPad Prism v.9.5.0.
4. Conclusions
With increased patient cases, limited treatment diversity, and the complex and unclear pathogenesis of PD, the current state of research is far beyond a firm and near-complete understanding of the disease and, consequently, remains complicated with regards drug development. Current efforts for PD treatment continue to be symptomatic treatment and neuroprotection to delay its progression. Traditional medicines have been an excellent source of diverse compounds, and because of the complex mechanisms involved in PD, a multiple-targeted approach could potentially uncover interesting mechanisms and interactions. This study initially addressed the interaction of major RP metabolites for neuroprotective activities to Parkinson’s disease in silico, and emodin could be attributed as the main effect compound in the extracts. These metabolites inhibited Parkinson’s disease targets on par with controls and known inhibitors from its binding activity, and predicted the inhibition activity from structure–activity relationships with good chemical stability.
5. Future Directions
While there is an interesting outlook on the multi-targeted effect of RP metabolites on key PD targets, experimental analyses are required to validate the computational results from this study. A more thorough computational analysis (such as network pharmacology and molecular dynamics simulations) could be initially performed for the prediction of the mechanism of action. In addition to previously mentioned validation studies, the activity of RP extracts or decoctions, including such extracts with in vitro and in vivo models of PD, may provide additional insights into its treatment. Gene expression studies and protein–ligand interaction experiments are recommended for further interpretation of their mechanism of action. Following this, structure isolation and comparative explorations of these extracts should provide the structural moiety and molecular interactions necessary for PD treatment. Before advanced investigation and clinical trials, these compounds should be subjected to experimental pharmacokinetics and toxicity studies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms241813929/s1.
Author Contributions
Conceptualization; methodology; formal analysis; data curation; investigation, P.J.B.G., S.K.-H.H. and P.-W.T.; Resources, P.-W.T. and S.K.-H.H.; writing—original draft preparation, P.J.B.G.; writing—review and editing, K.A.D.C.-C., R.B.L., S.K.-H.H. and P.-W.T.; supervision, P.-W.T.; funding acquisition, S.K.-H.H. and P.-W.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research is funded by the Division of Urology, Department of Surgery, Chi Mei Medical Center (CMFHR111140), National Science and Technology Council, Taiwan (NSTC 112-2221-E-309-001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data files generated and presented in this study are available upon request of the corresponding author. Prediction of pharmacokinetic properties can be replicated using the webservers.
Acknowledgments
The authors sincerely appreciate the financial support from the Office of Research and Development, Chang Jung Christian University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chopade, P.; Chopade, N.; Zhao, Z.; Mitragotri, S.; Liao, R.; Chandran Suja, V. Alzheimer’s and Parkinson’s Disease Therapies in the Clinic. Bioeng. Transl. Med. 2023, 8, e10367. [Google Scholar] [CrossRef]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.H.T.P.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini-Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Ray Dorsey, E.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and Projected Future Economic Burden of Parkinson’s Disease in the U.S. NPJ Park. Dis. 2020, 6, 15. [Google Scholar] [CrossRef]
- Bhat, S.; Acharya, U.R.; Hagiwara, Y.; Dadmehr, N.; Adeli, H. Parkinson’s Disease: Cause Factors, Measurable Indicators, and Early Diagnosis. Comput. Biol. Med. 2018, 102, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Kim, S.; Seo, J.-H.; Suh, Y.-H. α-Synuclein, Parkinson’s Disease, and Alzheimer’s Disease. Parkinsonism Relat. Disord. 2004, 10, S9–S13. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [PubMed]
- Gnanaraj, C.; Sekar, M.; Fuloria, S.; Swain, S.S.; Gan, S.H.; Chidambaram, K.; Rani, N.N.I.M.; Balan, T.; Stephenie, S.; Lum, P.T.; et al. In Silico Molecular Docking Analysis of Karanjin against Alzheimer’s and Parkinson’s Diseases as a Potential Natural Lead Molecule for New Drug Design, Development and Therapy. Molecules 2022, 27, 2834. [Google Scholar] [CrossRef] [PubMed]
- Latif, S.; Jahangeer, M.; Maknoon Razia, D.; Ashiq, M.; Ghaffar, A.; Akram, M.; El Allam, A.; Bouyahya, A.; Garipova, L.; Ali Shariati, M.; et al. Dopamine in Parkinson’s Disease. Clin. Chim. Acta 2021, 522, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and Functional Basis of Dopamine Receptors and Their Role in Neurogenesis: Possible Implication for Parkinson’s Disease. J. Exp. Neurosci. 2018, 12, 117906951877982. [Google Scholar] [CrossRef]
- Takakusaki, K. Functional Neuroanatomy for Posture and Gait Control. J. Mov. Disord. 2017, 10, 1–17. [Google Scholar] [CrossRef]
- Rubin, J.E.; McIntyre, C.C.; Turner, R.S.; Wichmann, T. Basal Ganglia Activity Patterns in Parkinsonism and Computational Modeling of Their Downstream Effects. Eur. J. Neurosci. 2012, 36, 2213–2228. [Google Scholar] [CrossRef]
- Galvan, A.; Devergnas, A.; Wichmann, T. Alterations in Neuronal Activity in Basal Ganglia-Thalamocortical Circuits in the Parkinsonian State. Front. Neuroanat. 2015, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.-C.; Chen, Y.; Xu Yang ShenTu, C.-Y.; Peng, L.-H. Signaling Pathways in Parkinson’s Disease: Molecular Mechanisms and Therapeutic Interventions. Signal Transduct. Target. Ther. 2023, 8, 73. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative Stress in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, H.; Jost, W. Parkinson’s Disease: A Never Ending Story. J. Neural. Transm. 2023, 130, 735–736. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System. Cells 2022, 11, 2607. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of Mitochondrial Complex I Induces Progressive Parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 Mutations in Autosomal Dominant Late-Onset Parkinson’s Disease: A Genome-Wide Linkage and Sequencing Study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Battaglia, M.R.; Di Fazio, C.; Battaglia, S. Activated Tryptophan-Kynurenine Metabolic System in the Human Brain Is Associated with Learned Fear. Front. Mol. Neurosci. 2023, 16, 1217090. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Bhatia, S.; Al-Harrasi, A.; Zengin, G.; Bumbu, A.G.; Andronie-Cioara, F.L.; Nechifor, A.C.; et al. The Footprint of Kynurenine Pathway in Neurodegeneration: Janus-Faced Role in Parkinson’s Disorder and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 6737. [Google Scholar] [CrossRef]
- Bohár, Z.; Toldi, J.; Fülöp, F.; Vécsei, L. Changing the Face of Kynurenines and Neurotoxicity: Therapeutic Considerations. Int. J. Mol. Sci. 2015, 16, 9772–9793. [Google Scholar] [CrossRef]
- Arias-Vera, J. Abnormalities in Blood Pressure Regulation in a Patient with Parkinson’s Disease. Am. J. Hypertens. 2003, 16, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, H.; Noda, K.; Mitsui, R.; Hashitani, H. Role of Enteric Dopaminergic Neurons in Regulating Peristalsis of Rat Proximal Colon. Neurogastroenterol. Motil. 2021, 33, e14127. [Google Scholar] [CrossRef]
- Bogdanov, V.; Kim, A.; Nodel, M.; Pavlenko, T.; Pavlova, E.; Blokhin, V.; Chesnokova, N.; Ugrumov, M. A Pilot Study of Changes in the Level of Catecholamines and the Activity of α-2-Macroglobulin in the Tear Fluid of Patients with Parkinson’s Disease and Parkinsonian Mice. Int. J. Mol. Sci. 2021, 22, 4736. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Finberg, J.P.M. Inhibitors of MAO-B and COMT: Their Effects on Brain Dopamine Levels and Uses in Parkinson’s Disease. J. Neural Transm. 2019, 126, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A. Adenosine A2A Receptor Antagonists in Parkinson’s Disease: Progress in Clinical Trials from the Newly Approved Istradefylline to Drugs in Early Development and Those Already Discontinued. CNS Drugs 2014, 28, 455–474. [Google Scholar] [CrossRef]
- Kulisevsky, J.; Poyurovsky, M. Adenosine A2A-Receptor Antagonism and Pathophysiology of Parkinson’s Disease and Drug-Induced Movement Disorders. Eur. Neurol. 2012, 67, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Pillay, V.; Choonara, Y.E. Advances in the Treatment of Parkinson’s Disease. Prog. Neurobiol. 2007, 81, 29–44. [Google Scholar] [CrossRef]
- Sengupta, T.; Vinayagam, J.; Singh, R.; Jaisankar, P.; Mohanakumar, K.P. Plant-Derived Natural Products for Parkinson’s Disease Therapy. In Advances in Neurobiology; Springer: New York, NY, USA, 2016; Volume 12, pp. 415–496. [Google Scholar]
- Rahman, M.M.; Wang, X.; Islam, M.R.; Akash, S.; Supti, F.A.; Mitu, M.I.; Harun-Or-Rashid, M.; Aktar, M.N.; Khatun Kali, M.S.; Jahan, F.I.; et al. Multifunctional Role of Natural Products for the Treatment of Parkinson’s Disease: At a Glance. Front. Pharmacol. 2022, 13, 976385. [Google Scholar] [CrossRef]
- Khattak, A.K.; Hassan, S.M.; Mughal, S.S. General Overview of Phytochemistry and Pharmacological Potential of Rheum Palmatum (Chinese Rhubarb). Innovare J. Ayurvedic Sci. 2020, 8, 5–9. [Google Scholar] [CrossRef]
- Tsai, P.-W.; Hsieh, C.-Y.; Ting, J.U.; Rogio, K.G.G.; Lee, C.-J.; De Castro-Cruz, K.A.; Ciou, Y.-R.; Lien, T.-K.; Yang, L.-L.; Hsueh, C.-C.; et al. Unraveling the Bioenergy Production and Electron Transport Characteristics of Processed Rheum palmatum L. for Antiviral Drug Development. Ind. Crops Prod. 2023, 195, 116488. [Google Scholar] [CrossRef]
- Chen, B.-Y.; Lin, Y.-H.; Wu, Y.-C.; Hsueh, C.-C. Deciphering Electron-Shuttling Characteristics of Neurotransmitters to Stimulate Bioelectricity-Generating Capabilities in Microbial Fuel Cells. Appl. Biochem. Biotechnol. 2020, 191, 59–73. [Google Scholar] [CrossRef]
- Chen, B.-Y.; Wu, Y.-C.; Lin, Y.-H.; Tayo, L.L.; Tacas, A.C.; Hsueh, C.-C. Deciphering Electron-Shuttling Characteristics of Parkinson’s Disease Medicines via Bioenergy Extraction in Microbial Fuel Cells. Ind. Eng. Chem. Res. 2020, 59, 17124–17136. [Google Scholar] [CrossRef]
- Mitra, S.; Anjum, J.; Muni, M.; Das, R.; Rauf, A.; Islam, F.; Bin Emran, T.; Semwal, P.; Hemeg, H.A.; Alhumaydhi, F.A.; et al. Exploring the Journey of Emodin as a Potential Neuroprotective Agent: Novel Therapeutic Insights with Molecular Mechanism of Action. Biomed. Pharmacother. 2022, 149, 112877. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Meng, J.; Jiang, K.; Lan, H.; Wang, Z.; Lin, M.; Li, W.; Guo, H.; Wei, Y.; Mu, Y. Improving Protein–Ligand Docking and Screening Accuracies by Incorporating a Scoring Function Correction Term. Brief. Bioinform. 2022, 23, bbac051. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Mor, D.E.; Ischiropoulos, H. The Convergence of Dopamine and α-Synuclein: Implications for Parkinson’s Disease. J. Exp. Neurosci. 2018, 12, 117906951876136. [Google Scholar] [CrossRef]
- Sousa, V.L.; Bellani, S.; Giannandrea, M.; Yousuf, M.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. α-Synuclein and Its A30P Mutant Affect Actin Cytoskeletal Structure and Dynamics. Mol. Biol. Cell 2009, 20, 3725–3739. [Google Scholar] [CrossRef]
- Olanow, C.W.; Brundin, P. Parkinson’s Disease and Alpha Synuclein: Is Parkinson’s Disease a Prion-Like Disorder? Mov. Disord. 2013, 28, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, R.; Schapira, A.H.V. Parkinson Disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Riederer, P.; Müller, T. Monoamine Oxidase-B Inhibitors in the Treatment of Parkinson’s Disease: Clinical–Pharmacological Aspects. J. Neural Transm. 2018, 125, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Freitas, R.; Schapira, M. A Systematic Analysis of Atomic Protein–Ligand Interactions in the PDB. Med. Chem. Commun. 2017, 8, 1970–1981. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute Electronegativity and Hardness: Application to Inorganic Chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Koopmans, T. Über Die Zuordnung von Wellenfunktionen Und Eigenwerten Zu Den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Janak, J.F. Proof That ∂E/∂ni = ε in Density-Functional Theory. Phys. Rev. B 1978, 18, 7165–7168. [Google Scholar] [CrossRef]
- Chandrakumar, K.; Pal, S. The Concept of Density Functional Theory Based Descriptors and Its Relation with the Reactivity of Molecular Systems: A Semi-Quantitative Study. Int. J. Mol. Sci. 2002, 3, 324–337. [Google Scholar] [CrossRef]
- Choudhary, V.; Bhatt, A.; Dash, D.; Sharma, N. DFT Calculations on Molecular Structures, HOMO–LUMO Study, Reactivity Descriptors and Spectral Analyses of Newly Synthesized Diorganotin(IV) 2-chloridophenylacetohydroxamate Complexes. J. Comput. Chem. 2019, 40, 2354–2363. [Google Scholar] [CrossRef]
- Gazquez, J.L.; Mendez, F. The Hard and Soft Acids and Bases Principle: An Atoms in Molecules Viewpoint. J. Phys. Chem. 1994, 98, 4591–4593. [Google Scholar] [CrossRef]
- Oliveri, V. Toward the Discovery and Development of Effective Modulators of α-Synuclein Amyloid Aggregation. Eur. J. Med. Chem. 2019, 167, 10–36. [Google Scholar] [CrossRef] [PubMed]
- Park, H.R.; Kim, J.; Kim, T.; Jo, S.; Yeom, M.; Moon, B.; Choo, I.H.; Lee, J.; Lim, E.J.; Park, K.D.; et al. Oxazolopyridines and Thiazolopyridines as Monoamine Oxidase B Inhibitors for the Treatment of Parkinson’s Disease. Bioorg. Med. Chem. 2013, 21, 5480–5487. [Google Scholar] [CrossRef] [PubMed]
- von Kleist, L.; Michaelis, S.; Bartho, K.; Graebner, O.; Schlief, M.; Dreger, M.; Schrey, A.K.; Sefkow, M.; Kroll, F.; Koester, H.; et al. Identification of Potential Off-Target Toxicity Liabilities of Catechol-O-Methyltransferase Inhibitors by Differential Competition Capture Compound Mass Spectrometry. J. Med. Chem. 2016, 59, 4664–4675. [Google Scholar] [CrossRef] [PubMed]
- Jörg, M.; Shonberg, J.; Mak, F.S.; Miller, N.D.; Yuriev, E.; Scammells, P.J.; Capuano, B. Novel Adenosine A2A Receptor Ligands: A Synthetic, Functional and Computational Investigation of Selected Literature Adenosine A2A Receptor Antagonists for Extending into Extracellular Space. Bioorg. Med. Chem. Lett. 2013, 23, 3427–3433. [Google Scholar] [CrossRef]
- Kong, L.D.; Cheng, C.H.K.; Tan, R.X. Inhibition of MAO A and B by Some Plant-Derived Alkaloids, Phenols and Anthraquinones. J. Ethnopharmacol. 2004, 91, 351–355. [Google Scholar] [CrossRef]
- Buchwald, P. Soft Drugs: Design Principles, Success Stories, and Future Perspectives. Expert Opin. Drug Metab. Toxicol. 2020, 16, 645–650. [Google Scholar] [CrossRef]
- Tian, H.; Ketkar, R.; Tao, P. ADMETboost: A Web Server for Accurate ADMET Prediction. J. Mol. Model. 2022, 28, 408. [Google Scholar] [CrossRef]
- Sakaeda, T.; Okamura, N.; Nagata, S.; Yagami, T.; Horinouichi, M.; Okumura, K.; Yamashita, F.; Hashida, M. Molecular and Pharmacokinetic Properties of 222 Commercially Available Oral Drugs in Humans. Biol. Pharm. Bull. 2001, 24, 935–940. [Google Scholar] [CrossRef]
- Gomtsyan, A. Heterocycles in Drugs and Drug Discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Keen, P. Effect of Binding to Plasma Proteins on the Distribution, Activity and Elimination of Drugs. In Concepts in Biochemical Pharmacology; Springer: Berlin/Heidelberg, Germany, 1971; pp. 213–233. [Google Scholar]
- Di, L. An Update on the Importance of Plasma Protein Binding in Drug Discovery and Development. Expert Opin. Drug Discov. 2021, 16, 1453–1465. [Google Scholar] [CrossRef]
- Chico, L.K.; Behanna, H.A.; Hu, W.; Zhong, G.; Roy, S.M.; Watterson, D.M. Molecular Properties and CYP2D6 Substrates: Central Nervous System Therapeutics Case Study and Pattern Analysis of a Substrate Database. Drug Metab. Dispos. 2009, 37, 2204–2211. [Google Scholar] [CrossRef]
- Dong, X.; Fu, J.; Yin, X.; Cao, S.; Li, X.; Lin, L.; Ni, J. Emodin: A Review of Its Pharmacology, Toxicity and Pharmacokinetics. Phytother. Res. 2016, 30, 1207–1218. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-Bound Human α-Synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef]
- Hubálek, F.; Binda, C.; Li, M.; Herzig, Y.; Sterling, J.; Youdim, M.B.H.; Mattevi, A.; Edmondson, D.E. Inactivation of Purified Human Recombinant Monoamine Oxidases A and B by Rasagiline and Its Analogues. J. Med. Chem. 2004, 47, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Hubálek, F.; Li, M.; Herzig, Y.; Sterling, J.; Edmondson, D.E.; Mattevi, A. Binding of Rasagiline-Related Inhibitors to Human Monoamine Oxidases: A Kinetic and Crystallographic Analysis. J. Med. Chem. 2005, 48, 8148–8154. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Le Trong, I.; Stenkamp, R.E.; Parson, W.W. Crystal Structures of Human 108V and 108M Catechol O-Methyltransferase. J. Mol. Biol. 2008, 380, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.-P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.T.; Lane, J.R.; IJzerman, A.P.; Stevens, R.C. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).