
Diabetic Endothelial Cell Glycogen Synthase Kinase 3β Activation Induces VCAM1 Ectodomain Shedding

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. High-Fat Diet Combined with Low-Dose Streptozotocin Induces an Insulin-Resistant, T2D Phenotype in Mice
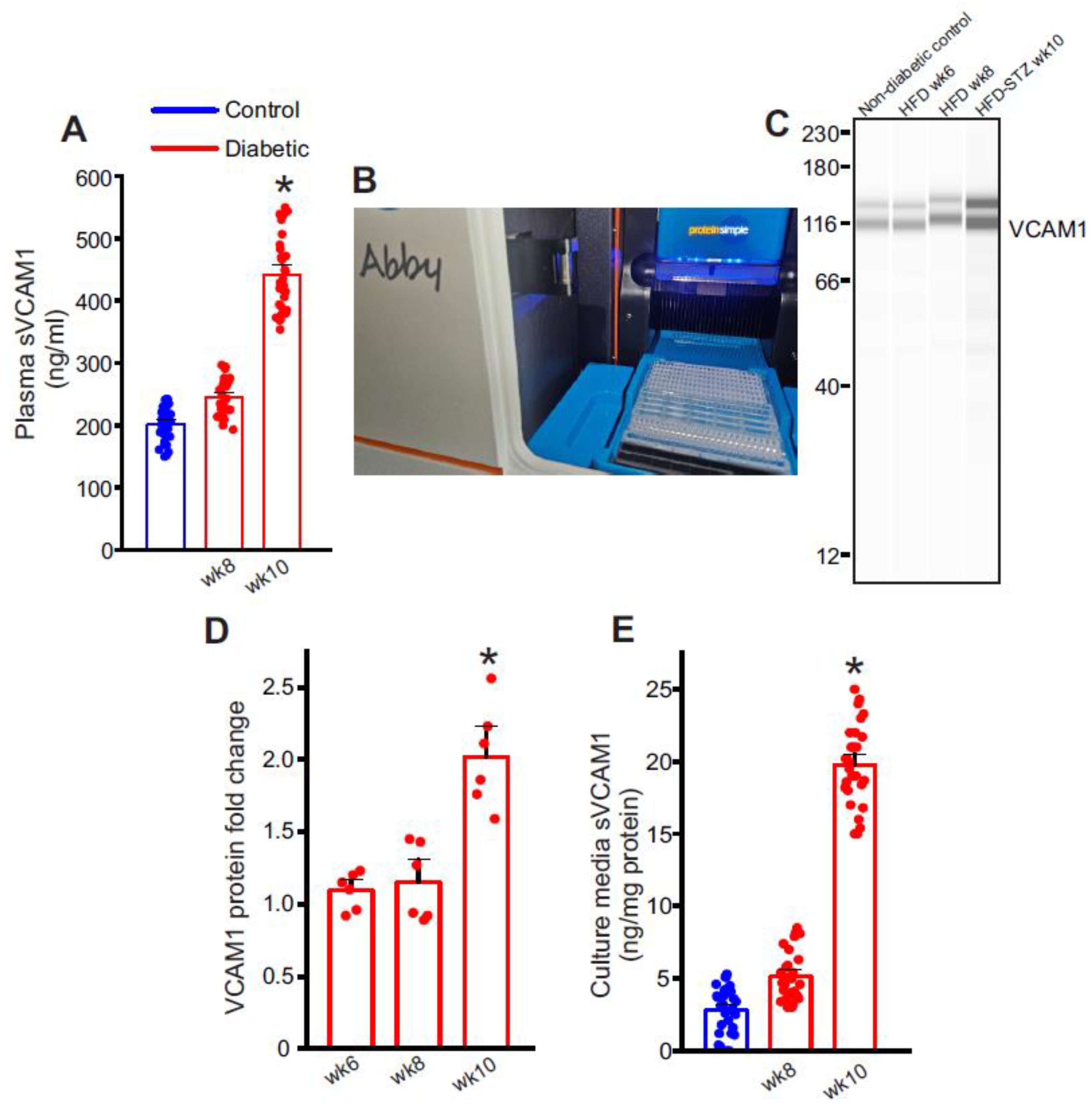
2.2. Plasma Soluble VCAM-1 (sVCAM1) Levels Increase with Insulin Resistance
2.3. Endothelial Cells as a Possible Source of Plasma sVCAM1 in Diabetic Animals
2.4. Diabetic Endothelial Cells Have Upregulated ADAM10 and ADAM17 Expression
2.5. Diabetic Endothelial Cell GSK3β Regulates the Expression of ADAM10/17 and VCAM1
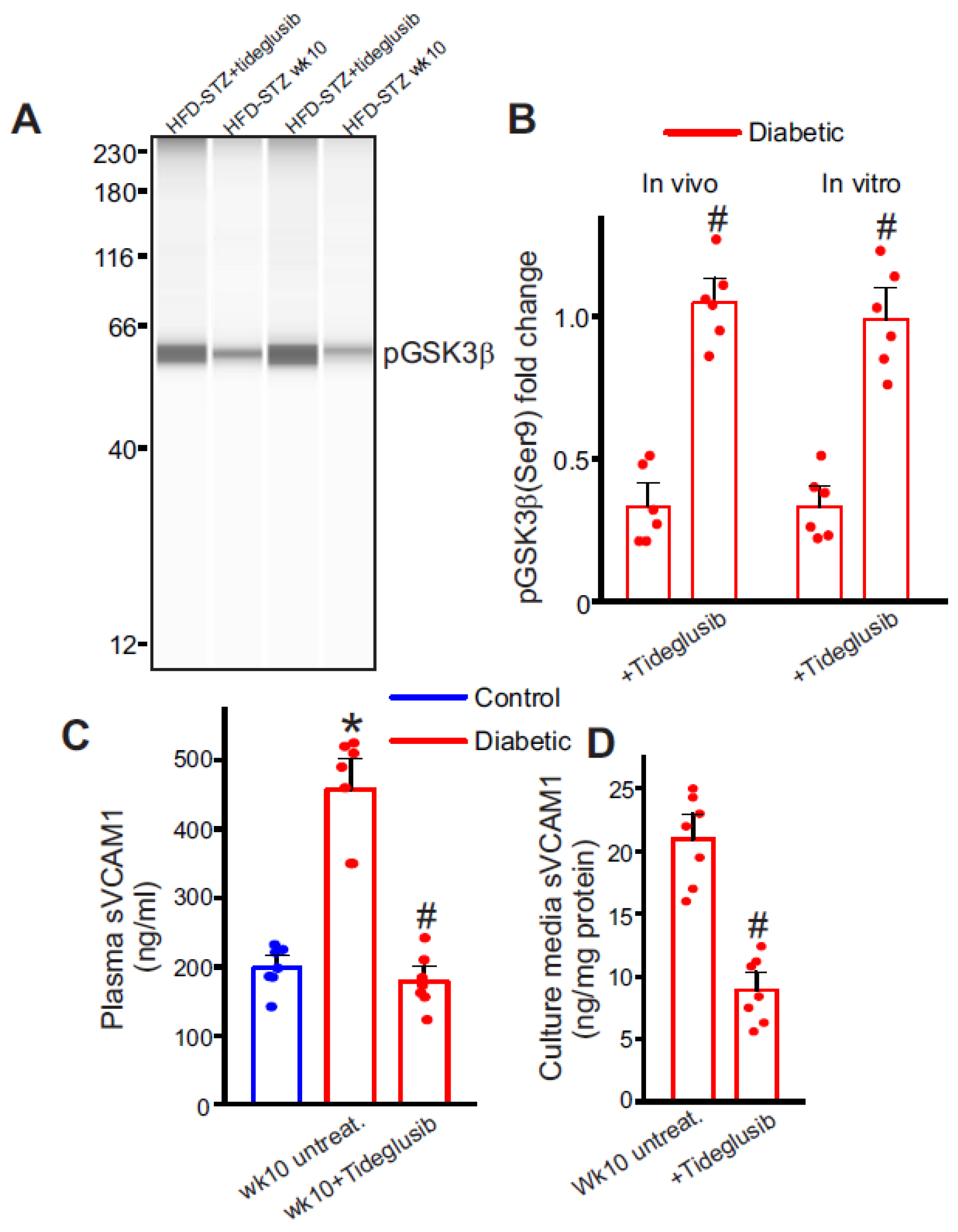
2.6. Selective Inhibition of GSK3β Downregulates Plasma sVCAM1 Levels
3. Discussion
4. Materials and Methods
4.1. Animal Usage
4.2. Tissue Preparation
4.3. Endothelial Cell (EC) Isolation and Culture
4.4. siRNA-Mediated Knockdown Studies
4.5. sVCAM1 ELISA
4.6. AbbyTM Capillary Electrophoresis Immunoassay (Simple Western)
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maric-Bilkan, C. Sex differences in micro- and macro-vascular complications of diabetes mellitus. Clin. Sci. 2017, 131, 833–846. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Harreiter, J. Sex and gender differences in therapy of type 2 diabetes. Diabetes Res. Clin. Pract. 2017, 131, 230–241. [Google Scholar] [CrossRef]
- Ramos, T.N.; Bullard, D.C.; Barnum, S.R. ICAM-1: Isoforms and Phenotypes. J. Immunol. 2014, 192, 4469. [Google Scholar] [CrossRef]
- Kallmann, B.A.; Hummel, V.; Toyka, K.V.; Rieckmann, P. Soluble VCAM-1 Release Indicates Inflammatory Blood-Brain Barrier Pathology and Further Modulates Adhesion. In Early Indicators Early Treatments Neuroprotection in Multiple Sclerosis; Hommes, O.R., Comi, G., Eds.; Topics in Neuroscience; Springer: Milan, Italy, 2004; pp. 115–117. [Google Scholar]
- Garton, K.J.; Gough, P.J.; Philalay, J.; Wille, P.T.; Blobel, C.P.; Whitehead, R.H.; Dempsey, P.J.; Raines, E.W. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J. Biol. Chem. 2003, 278, 37459–37464. [Google Scholar] [CrossRef]
- Bruno, C.M.; Valenti, M.; Bertino, G.; Ardiri, A.; Bruno, F.; Cunsolo, M.; Pulvirenti, D.; Neri, S. Plasma ICAM-1 and VCAM-1 levels in type 2 diabetic patients with and without microalbuminuria. Minerva Med. 2008, 99, 1–5. [Google Scholar]
- Troncoso, M.F.; Ortiz-Quintero, J.; Garrido-Moreno, V.; Sanhueza-Olivares, F.; Guerrero-Moncayo, A.; Chiong, M.; Castro, P.F.; García, L.; Gabrielli, L.; Corbalán, R.; et al. VCAM-1 as a predictor biomarker in cardiovascular disease. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2021, 1867, 166170. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Crandall, J.; Hori, O.; Cao, R.; Lakatta, E. Elevated plasma levels of vascular cell adhesion molecule-1 (VCAM-1) in diabetic patients with microalbuminuria: A marker of vascular dysfunction and progressive vascular disease. Br. J. Haematol. 1996, 92, 747–750. [Google Scholar] [CrossRef]
- Xu, Y.; Hou, H.; Zhao, L. The role of VCAM-1 in diabetic retinopathy: A systematic review and meta-analysis. J. Diabetes Its Complicat. 2023, 37, 108380. [Google Scholar] [CrossRef]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef]
- Gustavsson, C.; Agardh, C.D.; Zetterqvist, A.V.; Nilsson, J.; Agardh, E.; Gomez, M.F. Vascular cellular adhesion molecule-1 (VCAM-1) expression in mice retinal vessels is affected by both hyperglycemia and hyperlipidemia. PLoS ONE 2010, 5, e12699. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Q.; He, J.; Li, Y. Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front. Immunol. 2022, 13, 958790. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, G.A.; Awan, Z.; Hashem, E.; Al-Ama, N.; Abunaji, A.B. Levels of soluble cell adhesion molecules in type 2 diabetes mellitus patients with macrovascular complications. J. Int. Med. Res. 2020, 48, 300060519893858. [Google Scholar] [CrossRef] [PubMed]
- Hocaoglu-Emre, F.S.; Saribal, D.; Yenmis, G.; Guvenen, G. Vascular Cell Adhesion Molecule 1, Intercellular Adhesion Molecule 1, and Cluster of Differentiation 146 Levels in Patients with Type 2 Diabetes with Complications. Endocrinol. Metab. 2017, 32, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Hackman, A.; Abe, Y.; Insull, W.; Pownall, H.; Smith, L.; Dunn, K.; Gotto, A.M.; Ballantyne, C.M. Levels of Soluble Cell Adhesion Molecules in Patients With Dyslipidemia. Circulation 1996, 93, 1334–1338. [Google Scholar] [CrossRef]
- Koenen, R.R.; Pruessmeyer, J.; Soehnlein, O.; Fraemohs, L.; Zernecke, A.; Schwarz, N.; Reiss, K.; Sarabi, A.; Lindbom, L.; Hackeng, T.M.; et al. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood 2009, 113, 4799–4809. [Google Scholar] [CrossRef]
- Tsakadze, N.L.; Sithu, S.D.; Sen, U.; English, W.R.; Murphy, G.; D’Souza, S.E. Tumor Necrosis Factor-α-converting Enzyme (TACE/ADAM-17) Mediates the Ectodomain Cleavage of Intercellular Adhesion Molecule-1 (ICAM-1)*. J. Biol. Chem. 2006, 281, 3157–3164. [Google Scholar] [CrossRef]
- Maas, S.L.; Donners, M.; van der Vorst, E.P.C. ADAM10 and ADAM17, Major Regulators of Chronic Kidney Disease Induced Atherosclerosis? Int. J. Mol. Sci. 2023, 24, 7309. [Google Scholar] [CrossRef]
- Leo, M.D.; Peixoto-Nieves, D.; Yin, W.; Raghavan, S.; Muralidharan, P.; Mata-Daboin, A.; Jaggar, J.H. TMEM16A channel upregulation in arterial smooth muscle cells produces vasoconstriction during diabetes. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1089–H1101. [Google Scholar] [CrossRef]
- Nagy, C.; Einwallner, E. Study of In Vivo Glucose Metabolism in High-fat Diet-fed Mice Using Oral Glucose Tolerance Test (OGTT) and Insulin Tolerance Test (ITT). J. Vis. Exp. 2018, 131, e56672. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Berardi, C.; Wassel, C.L.; Decker, P.A.; Larson, N.B.; Kirsch, P.S.; Andrade, M.; Tsai, M.Y.; Pankow, J.S.; Sale, M.M.; Sicotte, H.; et al. Elevated Levels of Adhesion Proteins Are Associated With Low Ankle-Brachial Index. Angiology 2017, 68, 322–329. [Google Scholar] [CrossRef]
- Kotteas, E.A.; Boulas, P.; Gkiozos, I.; Tsagkouli, S.; Tsoukalas, G.; Syrigos, K.N. The Intercellular Cell Adhesion Molecule-1 (ICAM-1) in Lung Cancer: Implications for Disease Progression and Prognosis. Anticancer Res. 2014, 34, 4665. [Google Scholar]
- Champagne, B.; Tremblay, P.; Cantin, A.; St. Pierre, Y. Proteolytic Cleavage of ICAM-1 by Human Neutrophil Elastase. J. Immunol. 1998, 161, 6398. [Google Scholar] [CrossRef]
- Witte, D.R.; Broekmans, W.M.; Kardinaal, A.F.; Klöpping-Ketelaars, I.A.; van Poppel, G.; Bots, M.L.; Kluft, C.; Princen, J.M. Soluble intercellular adhesion molecule 1 and flow-mediated dilatation are related to the estimated risk of coronary heart disease independently from each other. Atherosclerosis 2003, 170, 147–153. [Google Scholar] [CrossRef]
- Semaan, H.B.; Gurbel, P.A.; Anderson, J.L.; Muhlestein, J.B.; Carlquist, J.F.; Horne, B.D.; Serebruany, V.L. Soluble VCAM-1 and E-Selectin, but Not ICAM-1 Discriminate Endothelial Injury in Patients with Documented Coronary Artery Disease. Cardiology 2000, 93, 7–10. [Google Scholar] [CrossRef]
- Demerath, E.; Towne, B.; Blangero, J.; Siervogel, R.M. The relationship of soluble ICAM-1, VCAM-1, P-selectin and E-selectin to cardiovascular disease risk factors in healthy men and women. Ann. Hum. Biol. 2001, 28, 664–678. [Google Scholar] [CrossRef]
- Haarmann, A.; Nowak, E.; Deiß, A.; van der Pol, S.; Monoranu, C.M.; Kooij, G.; Müller, N.; van der Valk, P.; Stoll, G.; de Vries, H.E.; et al. Soluble VCAM-1 impairs human brain endothelial barrier integrity via integrin α-4-transduced outside-in signalling. Acta Neuropathol. 2015, 129, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Roitman-Johnson, B.; Stampfer, M.J.; Allen, J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet 1998, 351, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.J.; Ballantyne, C.M.; Sharrett, A.R.; Smith, L.C.; Davis, C.E.; Gotto, A.M., Jr.; Boerwinkle, E. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: The Atherosclerosis Risk In Communities (ARIC) study. Circulation 1997, 96, 4219–4225. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Nheu, L.; Komesaroff, P.A. Cell adhesion molecules as pharmaceutical target in atherosclerosis. Mini Rev. Med. Chem. 2012, 12, 175–183. [Google Scholar] [CrossRef]
- Allen, S.; Moran, N. Cell Adhesion Molecules: Therapeutic Targets for Inhibition of Inflammatory States. Semin. Thromb. Hemost. 2015, 41, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Liao, J.K. A mouse model of diet-induced obesity and insulin resistance. Methods Mol. Biol. 2012, 821, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, E.R.; Fu, Z.; Liu, D. Development of a nongenetic mouse model of type 2 diabetes. Exp. Diabetes Res. 2011, 2011, 416254. [Google Scholar] [CrossRef] [PubMed]
- Leiter, E.H. Multiple low-dose streptozotocin-induced hyperglycemia and insulitis in C57BL mice: Influence of inbred background, sex, and thymus. Proc. Natl. Acad. Sci. USA 1982, 79, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. 2021, 1, e78. [Google Scholar] [CrossRef]
- Leguina-Ruzzi, A.; Ortiz, R.; Velarde, V. The streptozotocin-high fat diet induced diabetic mouse model exhibits severe skin damage and alterations in local lipid mediators. Biomed. J. 2018, 41, 328–332. [Google Scholar] [CrossRef]
- Avtanski, D.; Pavlov, V.A.; Tracey, K.J.; Poretsky, L. Characterization of inflammation and insulin resistance in high-fat diet-induced male C57BL/6J mouse model of obesity. Anim. Model Exp. Med. 2019, 2, 252–258. [Google Scholar] [CrossRef]
- Parilla, J.H.; Willard, J.R.; Barrow, B.M.; Zraika, S. A Mouse Model of Beta-Cell Dysfunction as Seen in Human Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 6106051. [Google Scholar] [CrossRef]
- Nath, S.; Ghosh, S.K.; Choudhury, Y. A murine model of type 2 diabetes mellitus developed using a combination of high fat diet and multiple low doses of streptozotocin treatment mimics the metabolic characteristics of type 2 diabetes mellitus in humans. J. Pharmacol. Toxicol. Methods 2017, 84, 20–30. [Google Scholar] [CrossRef]
- Heydemann, A. An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2016, 2902351. [Google Scholar] [CrossRef] [PubMed]
- Mosser, R.E.; Maulis, M.F.; Moullé, V.S.; Dunn, J.C.; Carboneau, B.A.; Arasi, K.; Pappan, K.; Poitout, V.; Gannon, M. High-fat diet-induced β-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am. J. Physiol.-Endocrinol. Metab. 2015, 308, E573–E582. [Google Scholar] [CrossRef] [PubMed]
- Christians, J.K.; Lennie, K.I.; Wild, L.K.; Garcha, R. Effects of high-fat diets on fetal growth in rodents: A systematic review. Reprod. Biol. Endocrinol. 2019, 17, 39. [Google Scholar] [CrossRef]
- Lai, M.; Chandrasekera, P.C.; Barnard, N.D. You are what you eat, or are you? The challenges of translating high-fat-fed rodents to human obesity and diabetes. Nutr. Diabetes 2014, 4, e135. [Google Scholar] [CrossRef]
- Austin, G.L.; Ogden, L.G.; Hill, J.O. Trends in carbohydrate, fat, and protein intakes and association with energy intake in normal-weight, overweight, and obese individuals: 1971–2006. Am. J. Clin. Nutr. 2011, 93, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of HOMA modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef]
- Matthews, D.R. Insulin resistance and beta-cell function—A clinical perspective. Diabetes Obes. Metab. 2001, 3 (Suppl. S1), S28–S33. [Google Scholar] [CrossRef]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2013, 1832, 2216–2231. [Google Scholar] [CrossRef]
- Dreymueller, D.; Pruessmeyer, J.; Groth, E.; Ludwig, A. The role of ADAM-mediated shedding in vascular biology. Eur. J. Cell Biol. 2012, 91, 472–485. [Google Scholar] [CrossRef]
- Morsing, S.K.H.; Rademakers, T.; Brouns, S.L.N.; Stalborch, A.D.V.; Donners, M.; van Buul, J.D. ADAM10-Mediated Cleavage of ICAM-1 Is Involved in Neutrophil Transendothelial Migration. Cells 2021, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Palau, V.; Jarrín, J.; Villanueva, S.; Benito, D.; Márquez, E.; Rodríguez, E.; Soler, M.J.; Oliveras, A.; Gimeno, J.; Sans, L.; et al. Endothelial ADAM17 Expression in the Progression of Kidney Injury in an Obese Mouse Model of Pre-Diabetes. Int. J. Mol. Sci. 2021, 23, 221. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Nawaz, M.I.; Ahmad, A.; De Zutter, A.; Siddiquei, M.M.; Blanter, M.; Allegaert, E.; Gikandi, P.W.; De Hertogh, G.; Van Damme, J.; et al. Evaluation of Proteoforms of the Transmembrane Chemokines CXCL16 and CX3CL1, Their Receptors, and Their Processing Metalloproteinases ADAM10 and ADAM17 in Proliferative Diabetic Retinopathy. Front. Immunol. 2020, 11, 601639. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, L.; Thounaojam, M.; Tawfik, A.; Li, J.; Hussein, K.; Jahng, W.J.; Al-Shabrawey, M.; Kwok, H.F.; Bartoli, M.; Gutsaeva, D. Role of Endothelial ADAM17 in Early Vascular Changes Associated with Diabetic Retinopathy. J. Clin. Med. 2020, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Bandyopadhyay, G.; Standaert, M.L.; Galloway, L.; Moscat, J.; Farese, R.V. Evidence for involvement of protein kinase C (PKC)-zeta and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubes. Endocrinology 1997, 138, 4721–4731. [Google Scholar] [CrossRef]
- Standaert, M.L.; Galloway, L.; Karnam, P.; Bandyopadhyay, G.; Moscat, J.; Farese, R.V. Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J. Biol. Chem. 1997, 272, 30075–30082. [Google Scholar] [CrossRef]
- Eto, M.; Kouroedov, A.; Cosentino, F.; Lüscher, T.F. Glycogen synthase kinase-3 mediates endothelial cell activation by tumor necrosis factor-alpha. Circulation 2005, 112, 1316–1322. [Google Scholar] [CrossRef]
- del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andres, M.V.; Gomez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; Leon, T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimer’s Dis. JAD 2013, 33, 205–215. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Tolosa, E.; Litvan, I.; Hoglinger, G.U.; Burn, D.; Lees, A.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; Del Ser, T. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 470–478. [Google Scholar] [CrossRef]
- Ghiarone, T.; Castorena-Gonzalez, J.A.; Foote, C.A.; Ramirez-Perez, F.I.; Ferreira-Santos, L.; Cabral-Amador, F.J.; de la Torre, R.; Ganga, R.R.; Wheeler, A.A.; Manrique-Acevedo, C.; et al. ADAM17 cleaves the insulin receptor ectodomain on endothelial cells and causes vascular insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H688–H701. [Google Scholar] [CrossRef]
- MacKay, C.E.; Floen, M.; Leo, M.D.; Hasan, R.; Garrud, T.A.C.; Fernández-Peña, C.; Singh, P.; Malik, K.U.; Jaggar, J.H. A plasma membrane-localized polycystin-1/polycystin-2 complex in endothelial cells elicits vasodilation. eLife 2022, 11, e74765. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, J.L.; Walters, J.L.; Galliou, J.M.C.; Christian, R.J.; Qi, S.; Savenkova, M.I.; Ibarra, C.K.; Grogan, S.R.; Fuchs, R.A. Basolateral amygdala corticotropin-releasing factor receptor type 1 regulates context-cocaine memory strength during reconsolidation in a sex-dependent manner. Neuropharmacology 2021, 200, 108819. [Google Scholar] [CrossRef] [PubMed]
- Beekman, C.; Janson, A.A.; Baghat, A.; van Deutekom, J.C.; Datson, N.A. Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0195850. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Ali, S.; Gonzales, A.L.; Thakore, P.; Griffin, C.S.; Yamasaki, E.; Alvarado, M.G.; Johnson, M.T.; Trebak, M.; Earley, S. STIM1-dependent peripheral coupling governs the contractility of vascular smooth muscle cells. eLife 2022, 11, e70278. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brishti, M.A.; Raghavan, S.; Lamar, K.; Singh, U.P.; Collier, D.M.; Leo, M.D. Diabetic Endothelial Cell Glycogen Synthase Kinase 3β Activation Induces VCAM1 Ectodomain Shedding. Int. J. Mol. Sci. 2023, 24, 14105. https://doi.org/10.3390/ijms241814105
Brishti MA, Raghavan S, Lamar K, Singh UP, Collier DM, Leo MD. Diabetic Endothelial Cell Glycogen Synthase Kinase 3β Activation Induces VCAM1 Ectodomain Shedding. International Journal of Molecular Sciences. 2023; 24(18):14105. https://doi.org/10.3390/ijms241814105
Chicago/Turabian StyleBrishti, Masuma Akter, Somasundaram Raghavan, Kennedy Lamar, Udai P. Singh, Daniel M. Collier, and M. Dennis Leo. 2023. "Diabetic Endothelial Cell Glycogen Synthase Kinase 3β Activation Induces VCAM1 Ectodomain Shedding" International Journal of Molecular Sciences 24, no. 18: 14105. https://doi.org/10.3390/ijms241814105
APA StyleBrishti, M. A., Raghavan, S., Lamar, K., Singh, U. P., Collier, D. M., & Leo, M. D. (2023). Diabetic Endothelial Cell Glycogen Synthase Kinase 3β Activation Induces VCAM1 Ectodomain Shedding. International Journal of Molecular Sciences, 24(18), 14105. https://doi.org/10.3390/ijms241814105