
Endothelial Dysfunction in Cardiorenal Conditions: Implications of Endothelial Glucocorticoid Receptor-Wnt Signaling


Abstract
:1. Introduction
2. Cardiorenal Syndrome
3. Effects of Endothelial Dysfunction in Cardiorenal Syndrome
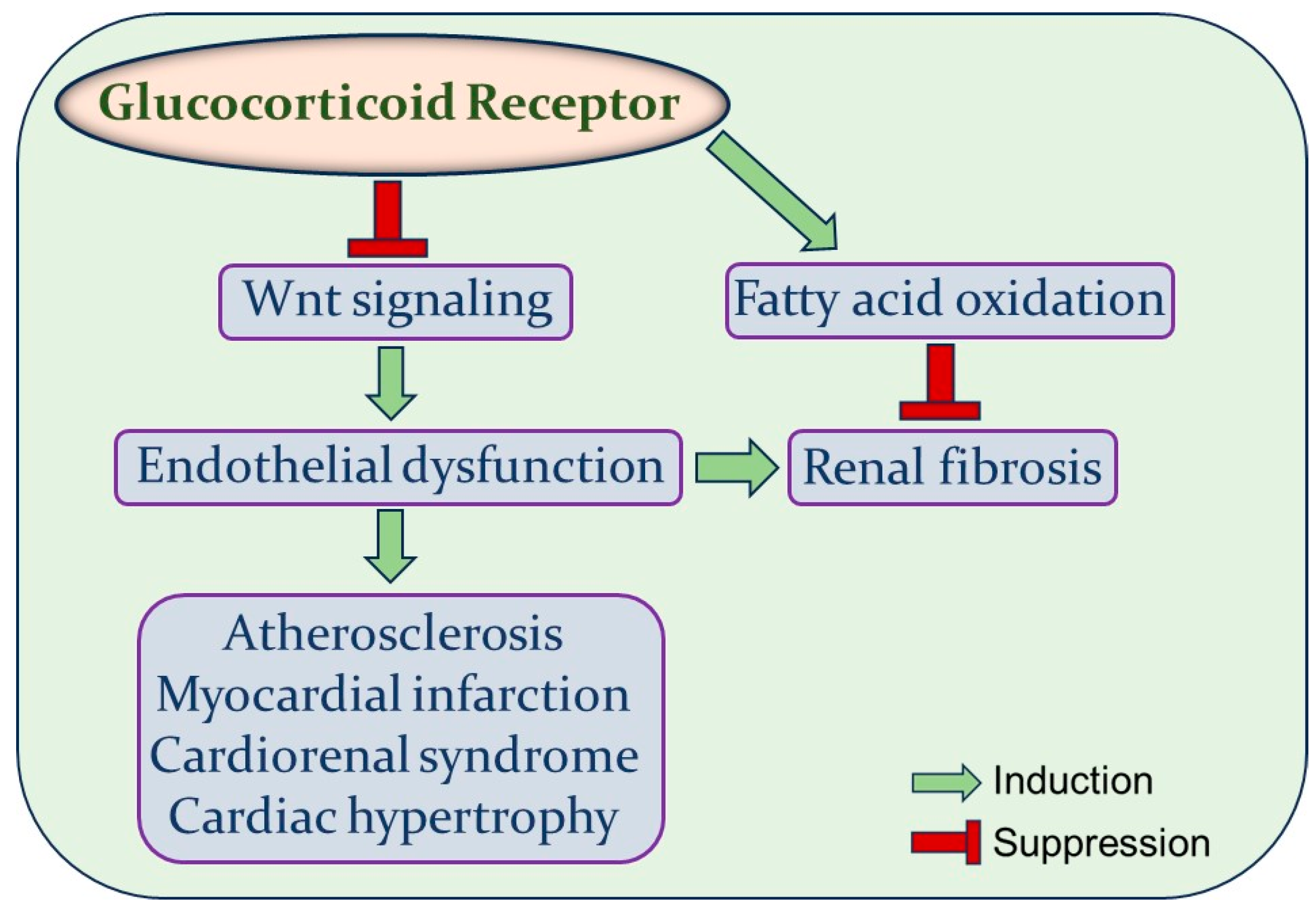
4. Glucocorticoid Receptor
5. Effects of GR on EC Metabolic Pathways
6. Endothelial GR and Wnt Signaling
7. Wnt Signaling in Cardiovascular Disease
7.1. Wnt Signaling in Atherosclerosis
7.2. Wnt Signaling in Myocardial Infarction
8. Wnt Signaling in Diabetic Nephropathy
9. Wnt Signaling in Cardiorenal Syndrome
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Bierhansl, L.; Conradi, L.C.; Treps, L.; Dewerchin, M.; Carmeliet, P. Central Role of Metabolism in Endothelial Cell Function and Vascular Disease. Physiology 2017, 32, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Maury, E.; Lehoux, S.; Guidet, B.; Offenstadt, G. The endothelium: Physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 2010, 36, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Thind, G.S.; Loehrke, M.; Wilt, J.L. Acute cardiorenal syndrome: Mechanisms and clinical implications. Cleve Clin. J. Med. 2018, 85, 231–239. [Google Scholar] [CrossRef] [PubMed]
- House, A.A.; Anand, I.; Bellomo, R.; Cruz, D.; Bobek, I.; Anker, S.D.; Aspromonte, N.; Bagshaw, S.; Berl, T.; Daliento, L.; et al. Definition and classification of Cardio-Renal Syndromes: Workgroup statements from the 7th ADQI Consensus Conference. Nephrol. Dial. Transplant. 2010, 25, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Yamagishi, S.; Yokoro, M.; Okuda, S. Role of asymmetric dimethylarginine in cardiorenal syndrome. Curr. Pharm. Des. 2014, 20, 2448–2455. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, N.W.; Nanayakkara, S.; Kaye, D.M. Pathogenesis and treatment of the cardiorenal syndrome: Implications of L-arginine-nitric oxide pathway impairment. Pharmacol. Ther. 2015, 154, 1–12. [Google Scholar] [CrossRef]
- Bertoluci, M.C.; Ce, G.V.; da Silva, A.M.; Wainstein, M.V.; Boff, W.; Punales, M. Endothelial dysfunction as a predictor of cardiovascular disease in type 1 diabetes. World J. Diabetes 2015, 6, 679–692. [Google Scholar] [CrossRef]
- Tomiyama, H.; Yamashina, A. Vascular Dysfunction: A Key Player in Chronic Cardio-renal Syndrome. Intern. Med. 2015, 54, 1465–1472. [Google Scholar] [CrossRef]
- Ueda, S.; Yamagishi, S.; Kaida, Y.; Okuda, S. Asymmetric dimethylarginine may be a missing link between cardiovascular disease and chronic kidney disease. Nephrology 2007, 12, 582–590. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E. Glucocorticoids and the Cardiovascular System. Adv. Exp. Med. Biol. 2015, 872, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Shimba, A.; Ikuta, K. Control of immunity by glucocorticoids in health and disease. Semin. Immunopathol. 2020, 42, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.N.; Wingenfeld, K.; Otte, C.; Meijer, O.C. Brain mineralocorticoid receptor in health and disease: From molecular signalling to cognitive and emotional function. Br. J. Pharmacol. 2022, 179, 3205–3219. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; Fletcher-Sananikone, E.; Sugimoto, H.; Hensel, J.; Lahiri, S.; Hertig, A.; Taduri, G.; Lawson, E.; Dewar, R.; Revuelta, I.; et al. Endothelial-to-mesenchymal transition compromises vascular integrity to induce Myc-mediated metabolic reprogramming in kidney fibrosis. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Bruning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid beta-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e813. [Google Scholar] [CrossRef]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; Garcia-Caballero, M.; Missiaen, R.; Huang, H.; Bruning, U.; et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 2017, 542, 49–54. [Google Scholar] [CrossRef]
- Lovisa, S.; Kalluri, R. Fatty Acid Oxidation Regulates the Activation of Endothelial-to-Mesenchymal Transition. Trends Mol. Med. 2018, 24, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Zhou, H.; Setia, O.; Liu, B.; Kanasaki, K.; Koya, D.; Dardik, A.; Fernandez-Hernando, C.; Goodwin, J. Loss of endothelial glucocorticoid receptor accelerates diabetic nephropathy. Nat. Commun. 2021, 12, 2368. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Dai, C.; Li, Y.; Zeng, G.; Monga, S.P.; Liu, Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2009, 20, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Sessa, W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc. Natl. Acad. Sci. USA 2013, 110, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Zhang, X.; Rotllan, N.; Feng, Y.; Zhou, H.; Fernandez-Hernando, C.; Yu, J.; Sessa, W.C. Endothelial glucocorticoid receptor suppresses atherogenesis-brief report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Mehta, S.; Srivastava, S.P.; Grabinska, K.; Zhang, X.; Wong, C.; Hedayat, A.; Perrotta, P.; Fernandez-Hernando, C.; Sessa, W.C.; et al. Endothelial cell-glucocorticoid receptor interactions and regulation of Wnt signaling. JCI Insight 2020, 5, e131384. [Google Scholar] [CrossRef] [PubMed]
- Boucher, P.; Matz, R.L.; Terrand, J. atherosclerosis: Gone with the Wnt? Atherosclerosis 2020, 301, 15–22. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Lu, Y.; Liu, X.; Huang, X.; Keller, E.T.; Qian, C.N.; Zhang, J. Wnt3a: Functions and implications in cancer. Chin. J. Cancer 2015, 34, 554–562. [Google Scholar] [CrossRef]
- Jang, J.; Jung, Y.; Chae, S.; Chung, S.I.; Kim, S.M.; Yoon, Y. WNT/beta-catenin pathway modulates the TNF-alpha-induced inflammatory response in bronchial epithelial cells. Biochem. Biophys. Res. Commun. 2017, 484, 442–449. [Google Scholar] [CrossRef]
- Manemann, S.M.; Gerber, Y.; Bielinski, S.J.; Chamberlain, A.M.; Margolis, K.L.; Weston, S.A.; Killian, J.M.; Roger, V.L. Recent trends in cardiovascular disease deaths: A state specific perspective. BMC Public. Health 2021, 21, 1031. [Google Scholar] [CrossRef]
- Hermans, K.C.; Blankesteijn, W.M. Wnt Signaling in Cardiac Disease. Compr. Physiol. 2015, 5, 1183–1209. [Google Scholar] [CrossRef] [PubMed]
- van de Schans, V.A.; Smits, J.F.; Blankesteijn, W.M. The Wnt/frizzled pathway in cardiovascular development and disease: Friend or foe? Eur. J. Pharmacol. 2008, 585, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, M.; Liew, C.C. Role of the adenomatous polyposis coli gene product in human cardiac development and disease. J. Biol. Chem. 2000, 275, 18470–18475. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Michael, A.; Andreucci, M.; Bhattacharya, K.; Dotto, P.; Walters, B.; Woodgett, J.; Kilter, H.; Force, T. Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4610–4615. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, C.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Zhou, L.; Liu, Y. An essential role for Wnt/beta-catenin signaling in mediating hypertensive heart disease. Sci. Rep. 2018, 8, 8996. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shevtsov, S.P.; Hsich, E.; Cui, L.; Haq, S.; Aronovitz, M.; Kerkela, R.; Molkentin, J.D.; Liao, R.; Salomon, R.N.; et al. The beta-catenin/T-cell factor/lymphocyte enhancer factor signaling pathway is required for normal and stress-induced cardiac hypertrophy. Mol. Cell. Biol. 2006, 26, 4462–4473. [Google Scholar] [CrossRef] [PubMed]
- Methatham, T.; Tomida, S.; Kimura, N.; Imai, Y.; Aizawa, K. Inhibition of the canonical Wnt signaling pathway by a beta-catenin/CBP inhibitor prevents heart failure by ameliorating cardiac hypertrophy and fibrosis. Sci. Rep. 2021, 11, 14886. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Barabutis, N.; Akhter, M.S.; Kubra, K.T.; Jackson, K. Growth Hormone-Releasing Hormone in Endothelial Inflammation. Endocrinology 2022, 164, bqac209. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Tsaousi, A.; Mill, C.; George, S.J. The Wnt pathways in vascular disease: Lessons from vascular development. Curr. Opin. Lipidol. 2011, 22, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Breton-Romero, R.; Feng, B.; Holbrook, M.; Farb, M.G.; Fetterman, J.L.; Linder, E.A.; Berk, B.D.; Masaki, N.; Weisbrod, R.M.; Inagaki, E.; et al. Endothelial Dysfunction in Human Diabetes Is Mediated by Wnt5a-JNK Signaling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Skaria, T.; Burgener, J.; Bachli, E.; Schoedon, G. IL-4 Causes Hyperpermeability of Vascular Endothelial Cells through Wnt5A Signaling. PLoS ONE 2016, 11, e0156002. [Google Scholar] [CrossRef] [PubMed]
- Christman, M.A., 2nd; Goetz, D.J.; Dickerson, E.; McCall, K.D.; Lewis, C.J.; Benencia, F.; Silver, M.J.; Kohn, L.D.; Malgor, R. Wnt5a is expressed in murine and human atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2864–H2870. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Kim, D.W.; Ha, Y.; Ihm, M.H.; Kim, H.; Song, K.; Lee, I. Wnt5a induces endothelial inflammation via beta-catenin-independent signaling. J. Immunol. 2010, 185, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Kim, C.S.; Naqvi, A.; Kumar, A.; Kumar, S.; Hoffman, T.A.; Irani, K. Epigenetic upregulation of p66shc mediates low-density lipoprotein cholesterol-induced endothelial cell dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H189–H196. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Gadi, I.; Nazir, S.; Al-Dabet, M.M.; Kohli, S.; Bock, F.; Breitenstein, L.; Ranjan, S.; Fuchs, T.; Halloul, Z.; et al. Activated protein C reverses epigenetically sustained p66(Shc) expression in plaque-associated macrophages in diabetes. Commun. Biol. 2018, 1, 104. [Google Scholar] [CrossRef]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Naqvi, A.; Hoffman, T.A.; Kumar, A.; Miller, F.J., Jr.; Kim, C.S.; Irani, K. Canonical Wnt signaling induces vascular endothelial dysfunction via p66Shc-regulated reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2301–2309. [Google Scholar] [CrossRef]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef]
- Cheng, S.L.; Shao, J.S.; Behrmann, A.; Krchma, K.; Towler, D.A. Dkk1 and MSX2-Wnt7b signaling reciprocally regulate the endothelial-mesenchymal transition in aortic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Di, M.; Wang, L.; Li, M.; Zhang, Y.; Liu, X.; Zeng, R.; Wang, H.; Chen, Y.; Chen, W.; Zhang, Y.; et al. Dickkopf1 destabilizes atherosclerotic plaques and promotes plaque formation by inducing apoptosis of endothelial cells through activation of ER stress. Cell Death Dis. 2017, 8, e2917. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, X.B.; Zhang, W.; Wu, L.D.; Liu, Y.S.; Hu, B.; Bi, C.L.; Chen, Y.F.; Liu, X.X.; Ge, C.; et al. Dickkopf-1 as a novel predictor is associated with risk stratification by GRACE risk scores for predictive value in patients with acute coronary syndrome: A retrospective research. PLoS ONE 2013, 8, e54731. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Otterdal, K.; Lekva, T.; Halvorsen, B.; Gabrielsen, A.; Sandberg, W.J.; Paulsson-Berne, G.; Pedersen, T.M.; Folkersen, L.; Gullestad, L.; et al. Dickkopf-1 enhances inflammatory interaction between platelets and endothelial cells and shows increased expression in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Baetta, R.; Banfi, C. Dkk (Dickkopf) Proteins. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, P.M.; Lewis, C.J.; House, D.L.; Keller, C.M.; Kohn, L.D.; Silver, M.J.; McCall, K.D.; Goetz, D.J.; Malgor, R. Increased Wnt5a mRNA Expression in Advanced Atherosclerotic Lesions, and Oxidized LDL Treated Human Monocyte-Derived Macrophages. Open Circ. Vasc. J. 2012, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.A.; Vengrenyuk, Y.; Menon, P.; Podolsky, I.; Feig, J.E.; Aderem, A.; Fisher, E.A.; Gold, E.S. Epigenome-guided analysis of the transcriptome of plaque macrophages during atherosclerosis regression reveals activation of the Wnt signaling pathway. PLoS Genet. 2014, 10, e1004828. [Google Scholar] [CrossRef]
- Weinstock, A.; Rahman, K.; Yaacov, O.; Nishi, H.; Menon, P.; Nikain, C.A.; Garabedian, M.L.; Pena, S.; Akbar, N.; Sansbury, B.E.; et al. Wnt signaling enhances macrophage responses to IL-4 and promotes resolution of atherosclerosis. Elife 2021, 10, e67932. [Google Scholar] [CrossRef]
- Albanese, I.; Khan, K.; Barratt, B.; Al-Kindi, H.; Schwertani, A. Atherosclerotic Calcification: Wnt Is the Hint. J. Am. Heart Assoc. 2018, 7, e007356. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.B.; Wang, W.E.; Zeng, C.Y. Wnt signaling pathways in myocardial infarction and the therapeutic effects of Wnt pathway inhibitors. Acta Pharmacol. Sin. 2019, 40, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Bastakoty, D.; Saraswati, S.; Joshi, P.; Atkinson, J.; Feoktistov, I.; Liu, J.; Harris, J.L.; Young, P.P. Temporary, Systemic Inhibition of the WNT/beta-Catenin Pathway promotes Regenerative Cardiac Repair following Myocardial Infarct. Cell Stem Cells Regen. Med. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model. Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, M.I.; Goumans, M.J.; van Middelaar, B.; Clevers, H.; Doevendans, P.A.; Sluijter, J.P. Active Wnt signaling in response to cardiac injury. Basic. Res. Cardiol. 2010, 105, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Zhou, H.; Zhang, L.S.; Tan, W.; Liu, Y.; Zhang, S.; Morlock, L.K.; Bao, X.; Palecek, S.P.; Feng, J.Q.; et al. Blockade to pathological remodeling of infarcted heart tissue using a porcupine antagonist. Proc. Natl. Acad. Sci. USA 2017, 114, 1649–1654. [Google Scholar] [CrossRef]
- Blumenthal, A.; Ehlers, S.; Lauber, J.; Buer, J.; Lange, C.; Goldmann, T.; Heine, H.; Brandt, E.; Reiling, N. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 2006, 108, 965–973. [Google Scholar] [CrossRef]
- Huang, L.; Xiang, M.; Ye, P.; Zhou, W.; Chen, M. Beta-catenin promotes macrophage-mediated acute inflammatory response after myocardial infarction. Immunol. Cell Biol. 2018, 96, 100–113. [Google Scholar] [CrossRef]
- Song, Z.; Wang, X.; He, L.; Chen, L.; Ren, Z.; Song, S. Suppression of lysosomal-associated protein transmembrane 5 ameliorates cardiac function and inflammatory response by inhibiting the nuclear factor-kappa B (NF-kappaB) pathway after myocardial infarction in mice. Exp. Anim. 2022, 71, 415–425. [Google Scholar] [CrossRef]
- Lin, J.C.; Chang, R.L.; Chen, Y.F.; Yang, J.J.; Baskaran, R.; Chung, L.C.; Chen, R.J.; Day, C.H.; Vijaya Padma, V.; Huang, C.Y. beta-Catenin overexpression causes an increase in inflammatory cytokines and NF-kappaB activation in cardiomyocytes. Cell. Mol. Biol. 2016, 63, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Almas, S.; Prabhakar, S. Wnt signaling and podocyte dysfunction in diabetic nephropathy. J. Investig. Med. 2017, 65, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, R.; Wu, X.; Chen, Y.; Ji, W.; Wang, J.; Zhang, Y.; Xia, Y.; Tang, Y.; Yuan, J. The Wnt Signaling Pathway in Diabetic Nephropathy. Front. Cell Dev. Biol. 2021, 9, 701547. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wen, J.; Dong, Y.; Zhang, Q.; Guan, J.; Liu, F.; Zhou, T.; Li, Z.; Fan, Y.; Wang, N. Wnt5a promotes renal tubular inflammation in diabetic nephropathy by binding to CD146 through noncanonical Wnt signaling. Cell Death Dis. 2021, 12, 92. [Google Scholar] [CrossRef]
- Dai, C.; Stolz, D.B.; Kiss, L.P.; Monga, S.P.; Holzman, L.B.; Liu, Y. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J. Am. Soc. Nephrol. 2009, 20, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Sun, Q.; Hua, M.R.; Suo, P.; Chen, J.R.; Yu, X.Y.; Zhao, Y.Y. Targeting the Wnt/beta-Catenin Signaling Pathway as a Potential Therapeutic Strategy in Renal Tubulointerstitial Fibrosis. Front. Pharmacol. 2021, 12, 719880. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.A.; Modarage, K.; Goggolidou, P. The Role of Wnt Signalling in Chronic Kidney Disease (CKD). Genes 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Garsen, M.; Rops, A.L.; Rabelink, T.J.; Berden, J.H.; van der Vlag, J. The role of heparanase and the endothelial glycocalyx in the development of proteinuria. Nephrol. Dial. Transplant. 2014, 29, 49–55. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; He, W.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Liu, Y. Mutual antagonism of Wilms’ tumor 1 and beta-catenin dictates podocyte health and disease. J. Am. Soc. Nephrol. 2015, 26, 677–691. [Google Scholar] [CrossRef]
- Maezawa, Y.; Takemoto, M.; Yokote, K. Cell biology of diabetic nephropathy: Roles of endothelial cells, tubulointerstitial cells and podocytes. J. Diabetes Investig. 2015, 6, 3–15. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Minto, A.W.; Wang, J.; Shi, Q.; Li, X.; Quigg, R.J. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J. 2008, 22, 4126–4135. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Hou, F.F.; Zhou, L.; Liu, Y. Wnt/beta-catenin signaling mediates both heart and kidney injury in type 2 cardiorenal syndrome. Kidney Int. 2019, 95, 815–829. [Google Scholar] [CrossRef]
- Zuo, Y.; Liu, Y. New insights into the role and mechanism of Wnt/beta-catenin signalling in kidney fibrosis. Nephrology 2018, 23 (Suppl. S4), 38–43. [Google Scholar] [CrossRef]

{kind=link}
| Phenotype | Nomenclature | Definition |
|---|---|---|
| Type 1 CRS | Acute CRS | Acute worsening of cardiac function leading to renal dysfunction. |
| Type 2 CRS | Chronic CRS | Chronic abnormalities in cardiac function leading to renal dysfunction. |
| Type 3 CRS | Acute reno- cardiac syndrome | Acute worsening of renal function causing cardiac dysfunction. |
| Type 4 CRS | Chronic reno- cardiac syndrome | Chronic abnormalities in renal function leading to cardiac disease. |
| Type 5 CRS | Secondary CRS | Systemic conditions causing simultaneous dysfunction of the heart and kidney. |
| Cell Types | Modulators | Molecular Pathways | Effects |
|---|---|---|---|
| Cardiomyocytes and cardiac fibroblasts | Angiotensin II | ↑ Wnt/β-catenin | Cardiac hypertrophy and myocardial fibrosis [35] |
| Endothelial cells | Wnt5a | ↑ Non-canonical Wnt pathway ↑ NF-κB pathway | Endothelial inflammation and atherosclerosis [46] |
| HUVECs | Wnt3a | ↑ Wnt/β-catenin | Endothelium oxidative stress and dysfunction [49] |
| Platelets and HUVECs | DKK-1 | ↑ NF-κB pathway ↓ Wnt/β-catenin | Endothelial dysfunction and atherosclerotic plaque formation [54] |
| Macrophages | ox-LDL | ↑ Non-canonical Wnt pathway | Expression of Wnt5a and progression of atherosclerosis [57] |
| Cardiac macrophages | Post MI | ↑ Wntβ-catenin ↑ NF-κB pathway | Macrophage-mediated inflammatory response after MI [69,70] |
| Podocytes | Adriamycin | ↑ Wnt/β-catenin | Podocyte dysfunction and albuminuria [80] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhter, M.S.; Goodwin, J.E. Endothelial Dysfunction in Cardiorenal Conditions: Implications of Endothelial Glucocorticoid Receptor-Wnt Signaling. Int. J. Mol. Sci. 2023, 24, 14261. https://doi.org/10.3390/ijms241814261
Akhter MS, Goodwin JE. Endothelial Dysfunction in Cardiorenal Conditions: Implications of Endothelial Glucocorticoid Receptor-Wnt Signaling. International Journal of Molecular Sciences. 2023; 24(18):14261. https://doi.org/10.3390/ijms241814261
Chicago/Turabian StyleAkhter, Mohammad Shohel, and Julie Elizabeth Goodwin. 2023. "Endothelial Dysfunction in Cardiorenal Conditions: Implications of Endothelial Glucocorticoid Receptor-Wnt Signaling" International Journal of Molecular Sciences 24, no. 18: 14261. https://doi.org/10.3390/ijms241814261
APA StyleAkhter, M. S., & Goodwin, J. E. (2023). Endothelial Dysfunction in Cardiorenal Conditions: Implications of Endothelial Glucocorticoid Receptor-Wnt Signaling. International Journal of Molecular Sciences, 24(18), 14261. https://doi.org/10.3390/ijms241814261