
Exploring Three Avenues: Chemo- and Regioselective Transformations of 1,2,4-Triketone Analogs into Pyrazoles and Pyridazinones
 , and
, and
Abstract
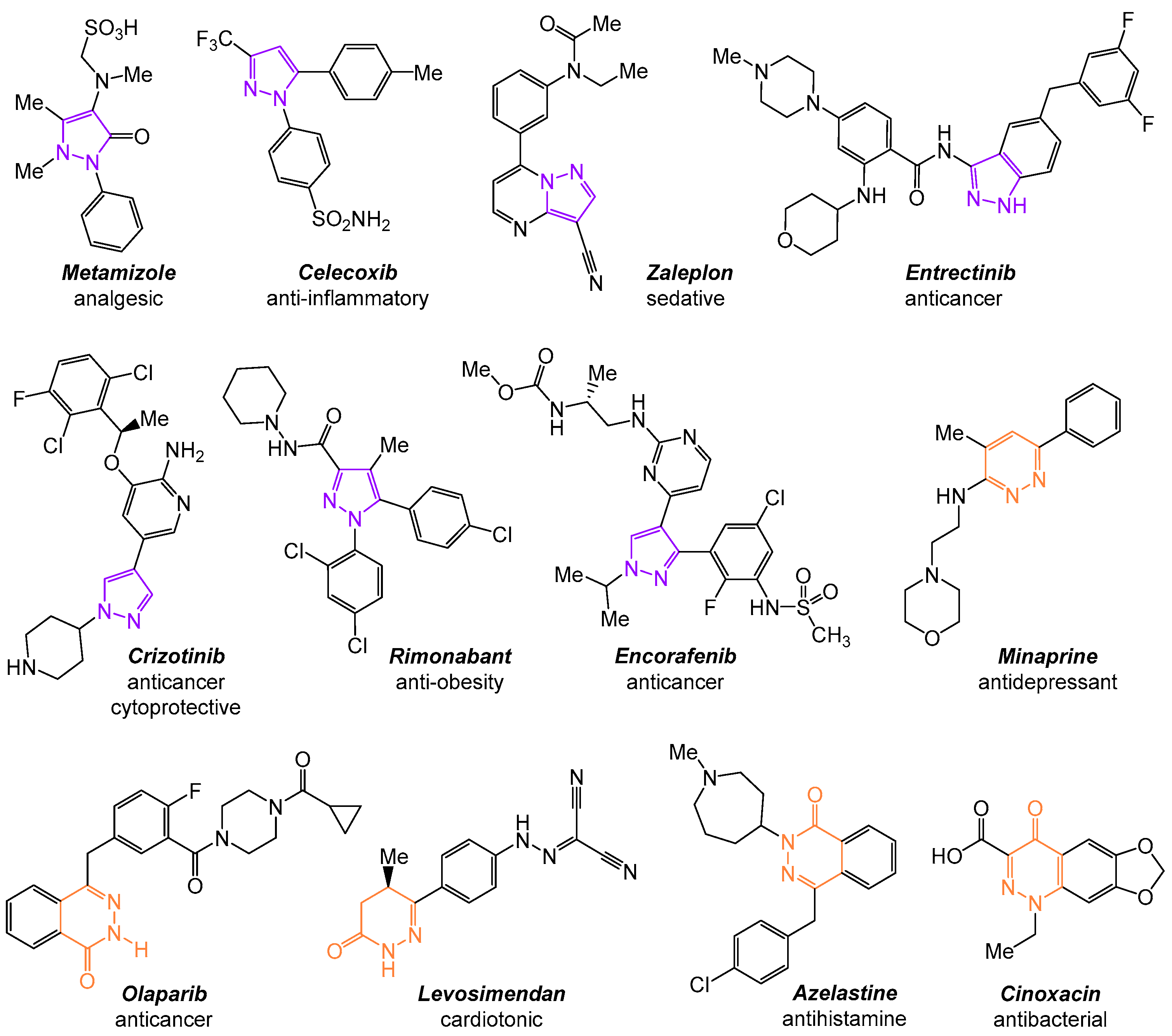
:1. Introduction
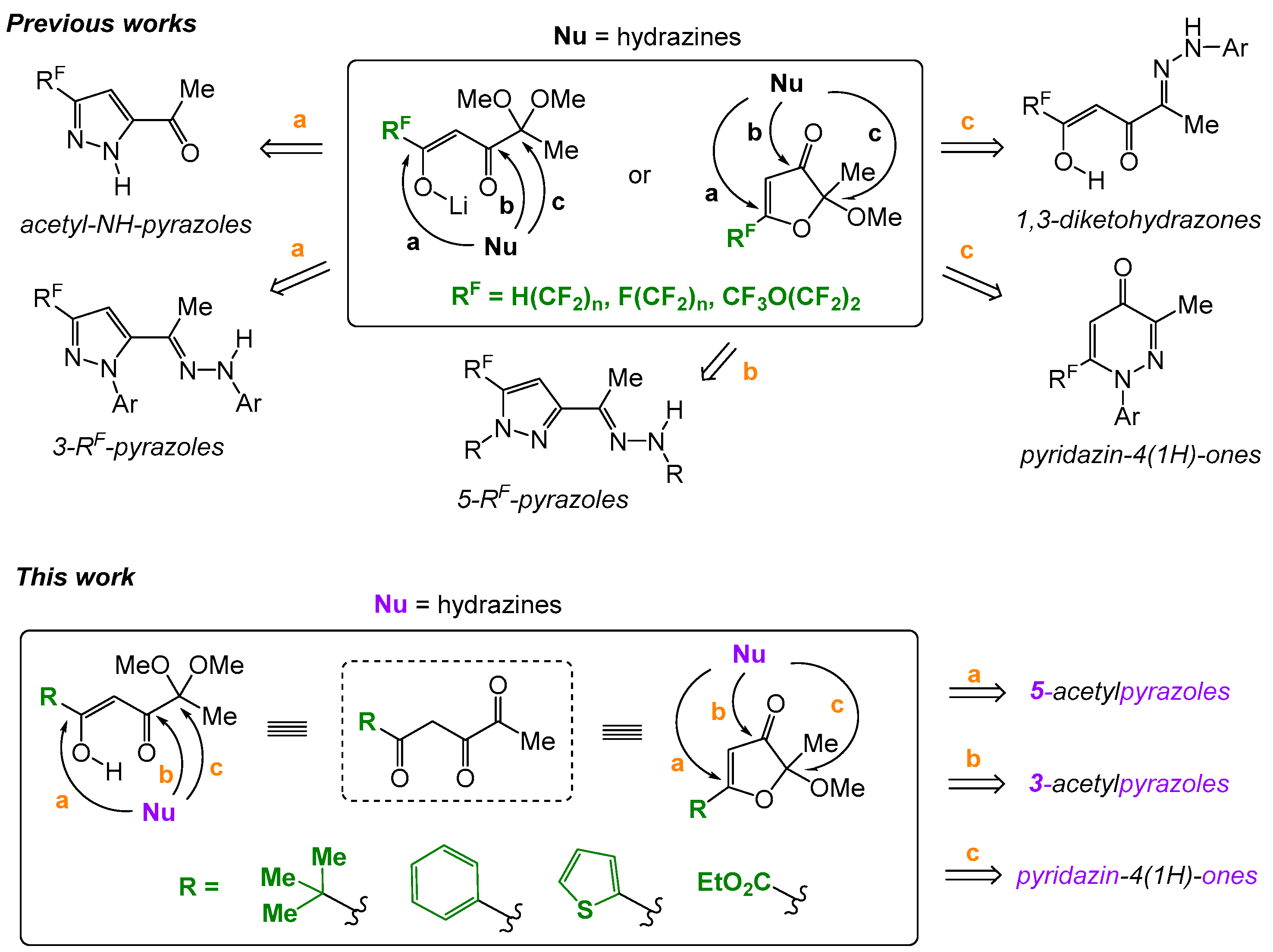
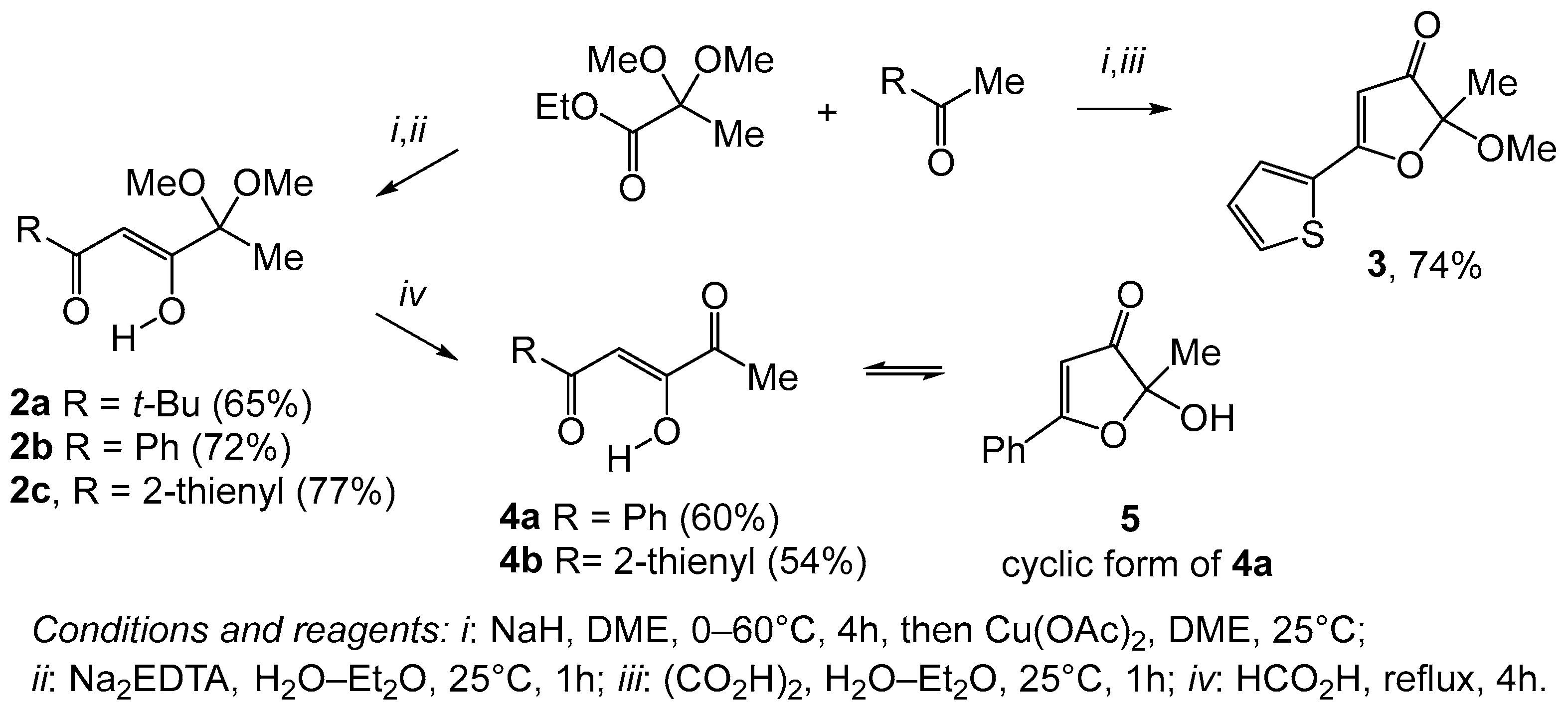
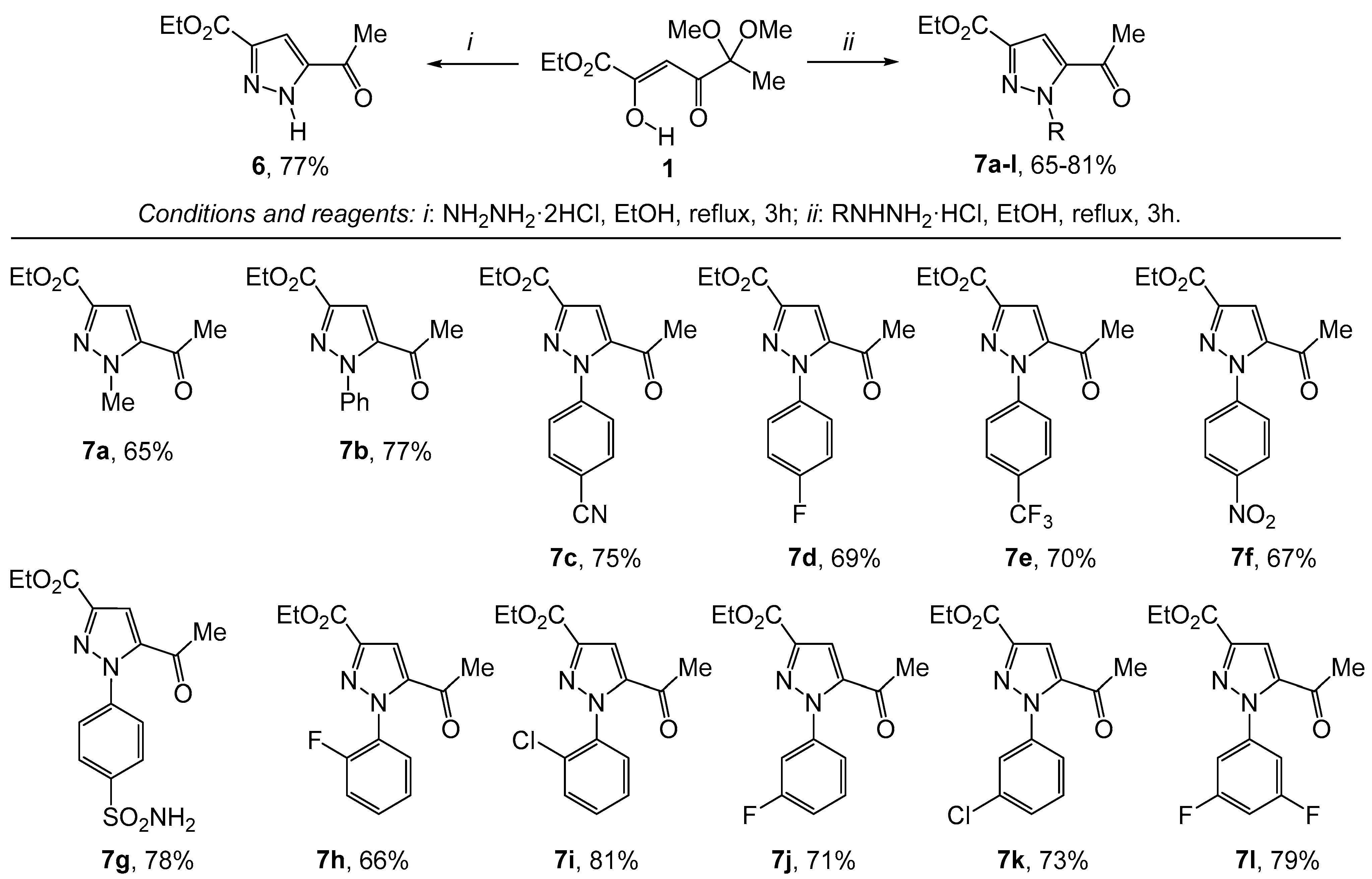
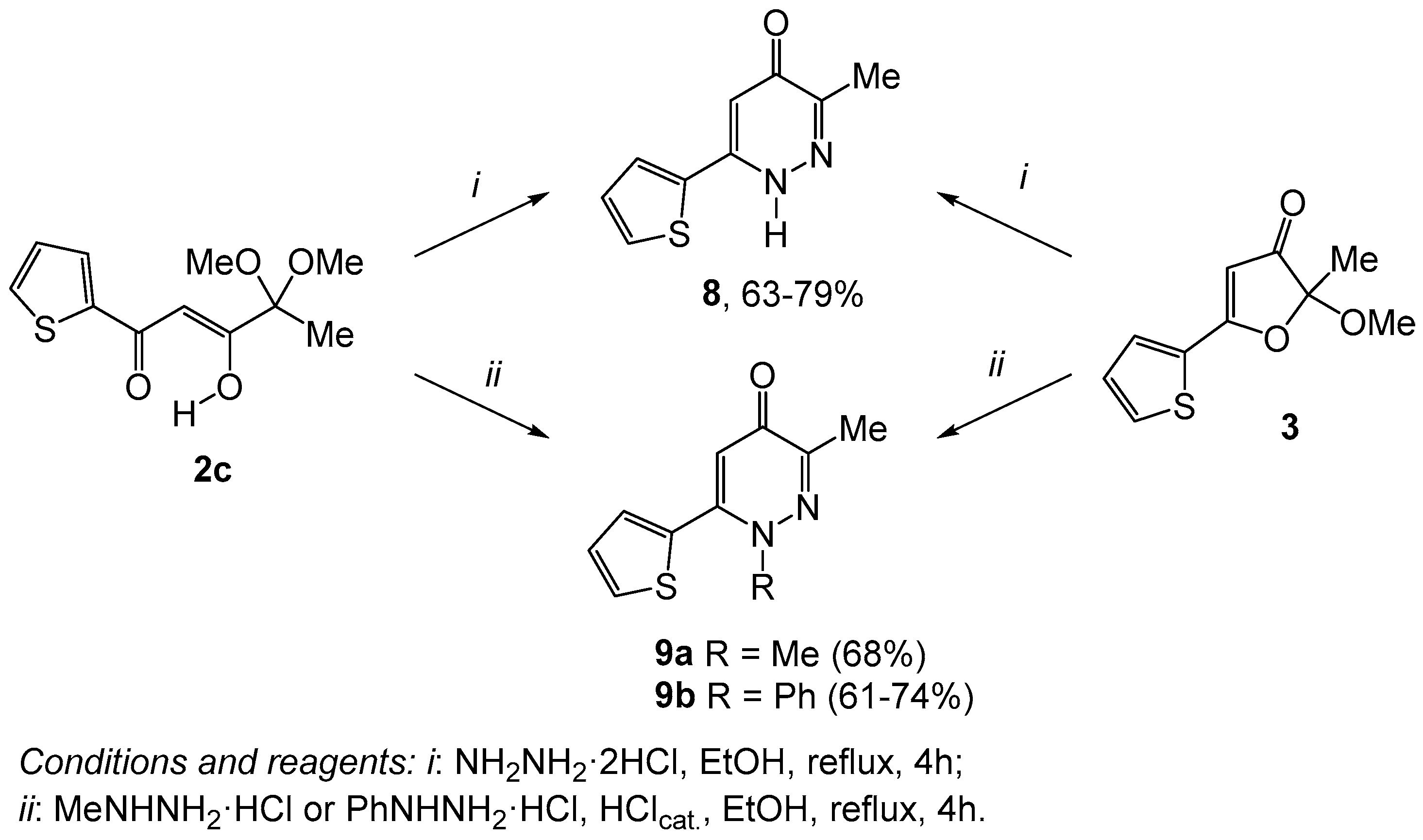
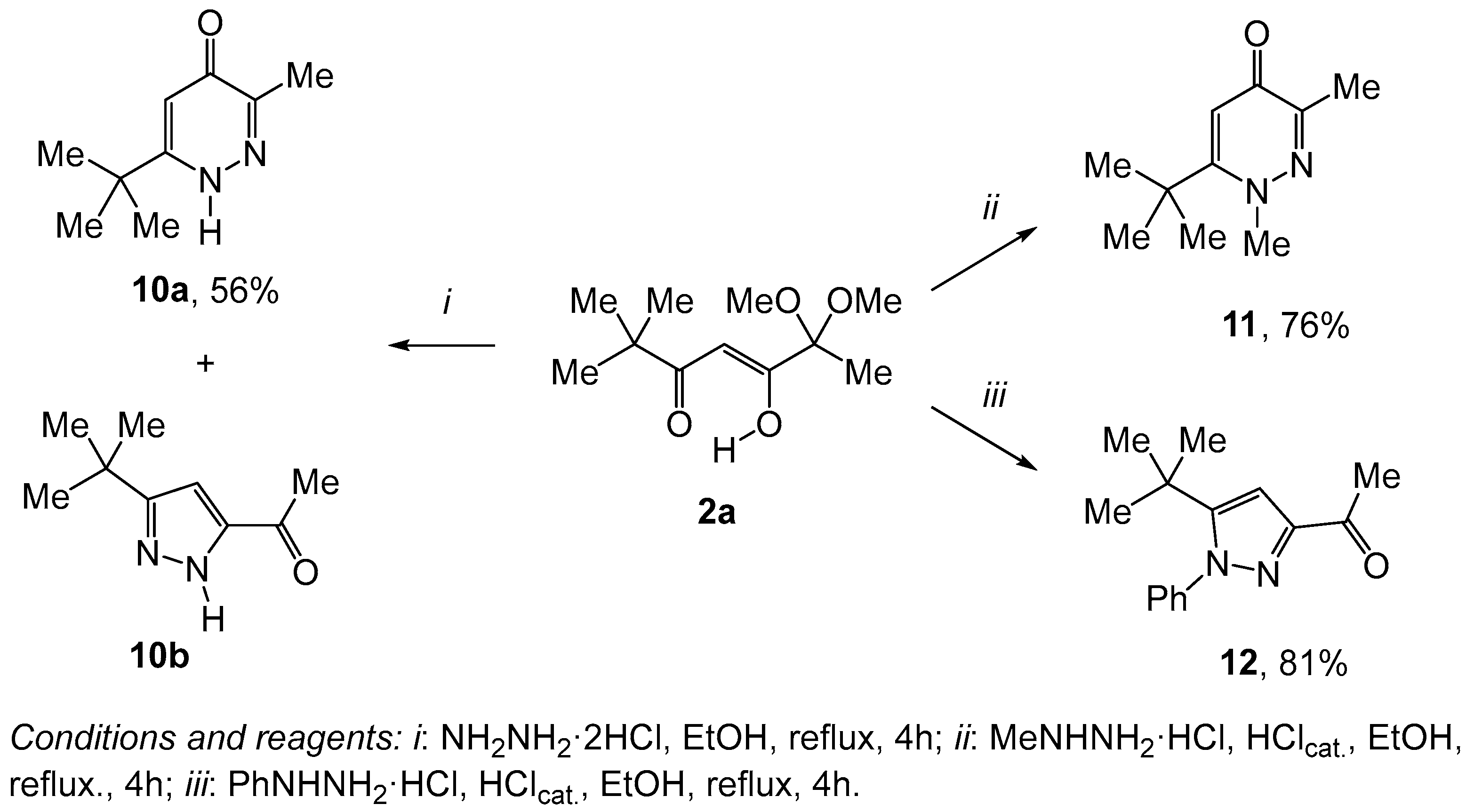
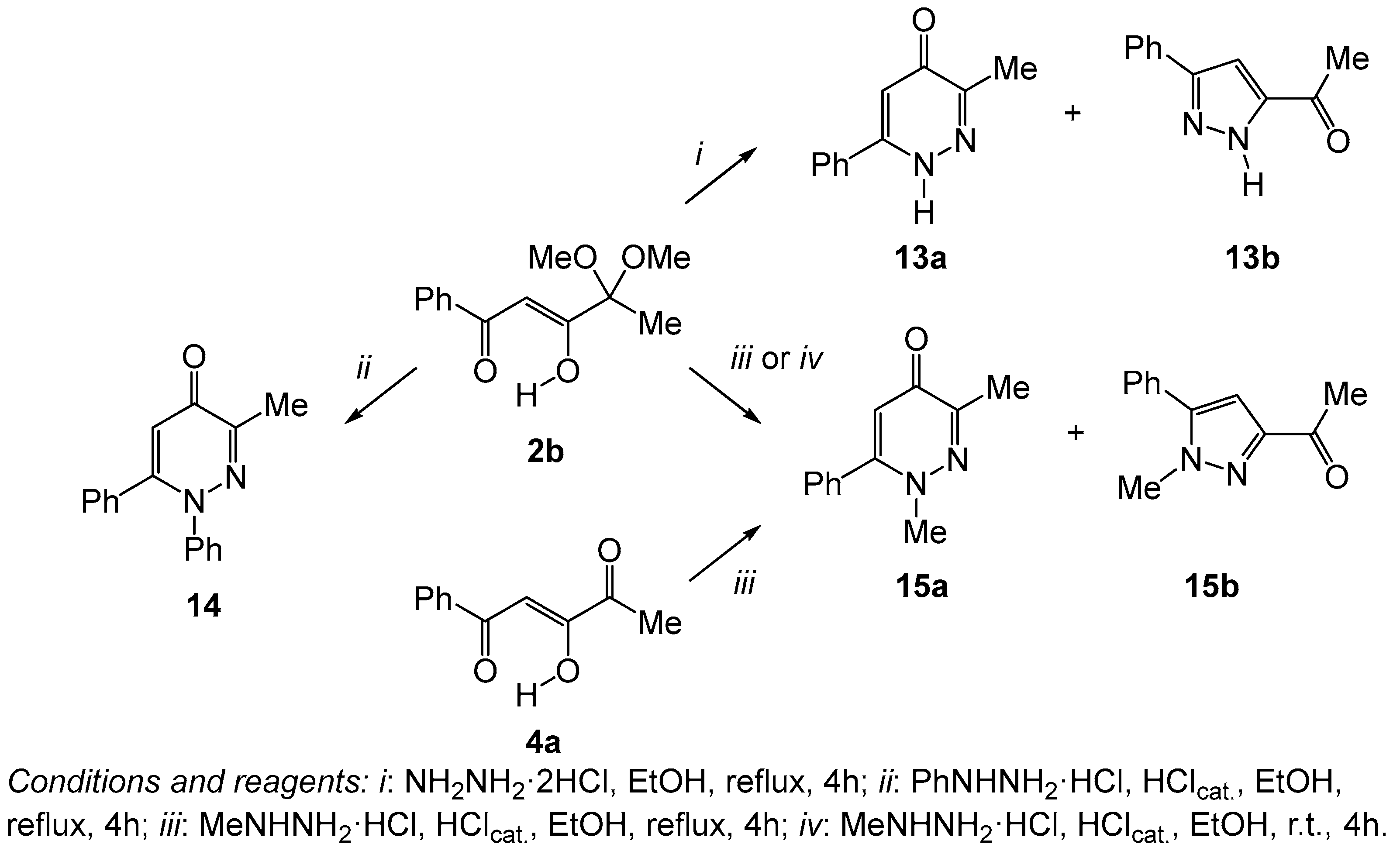
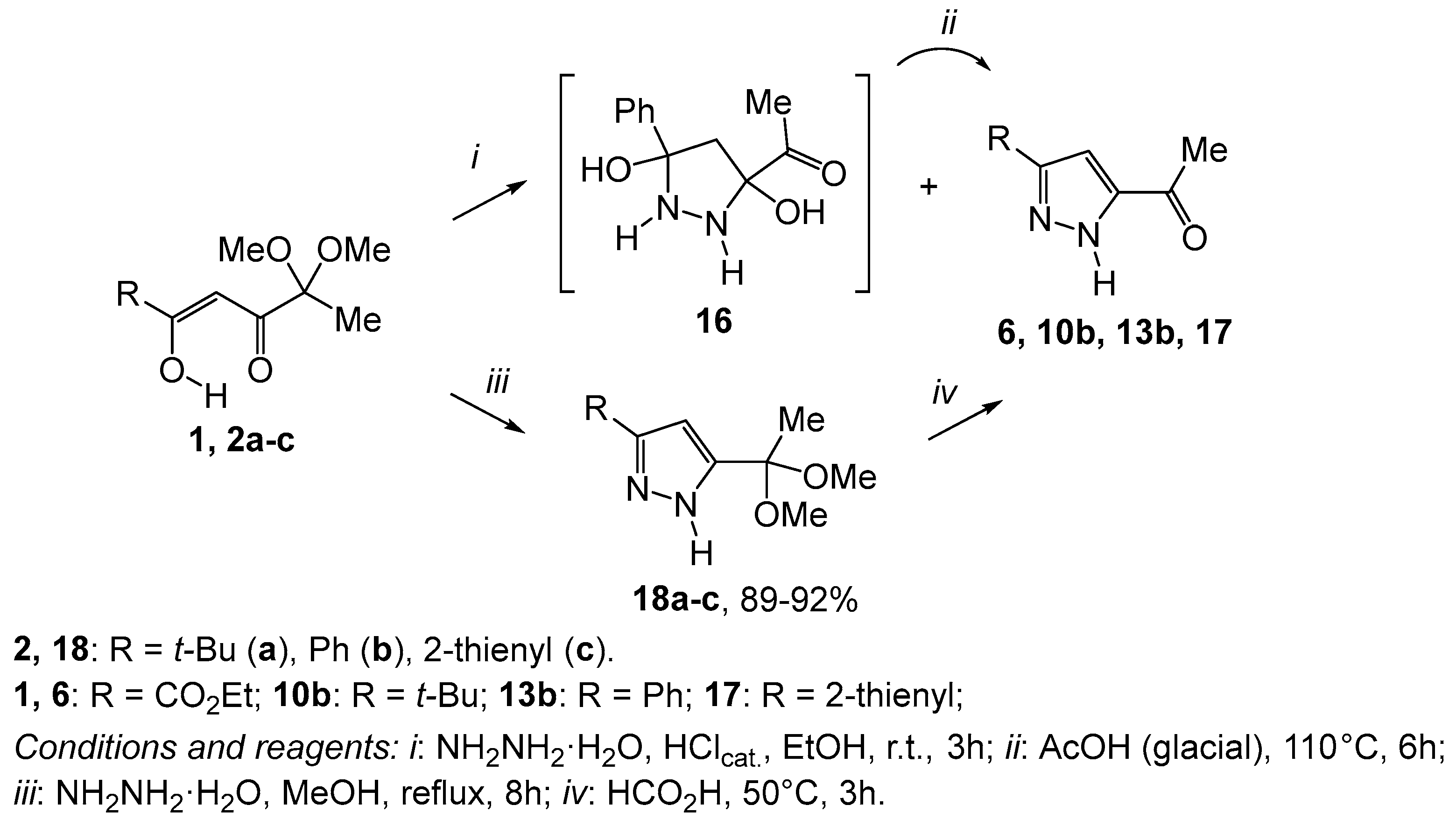
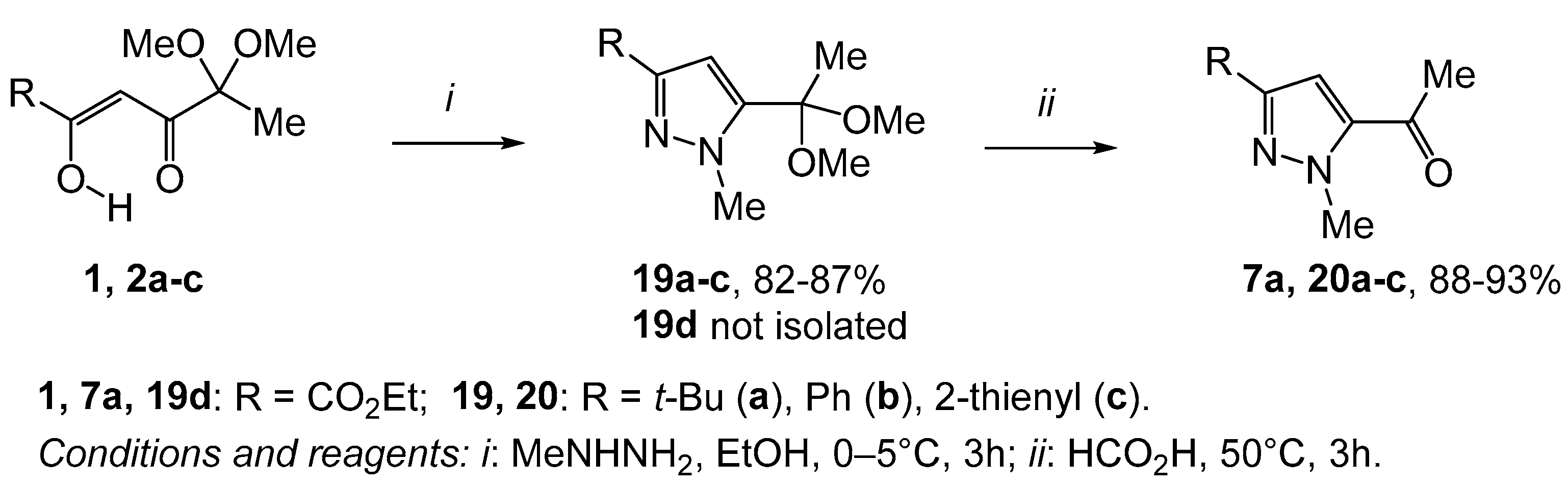
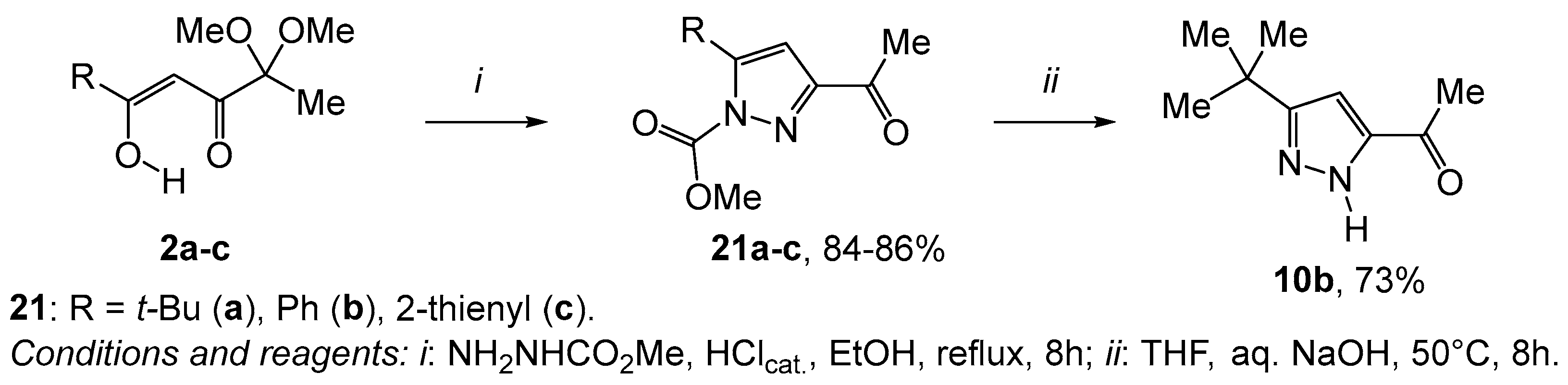
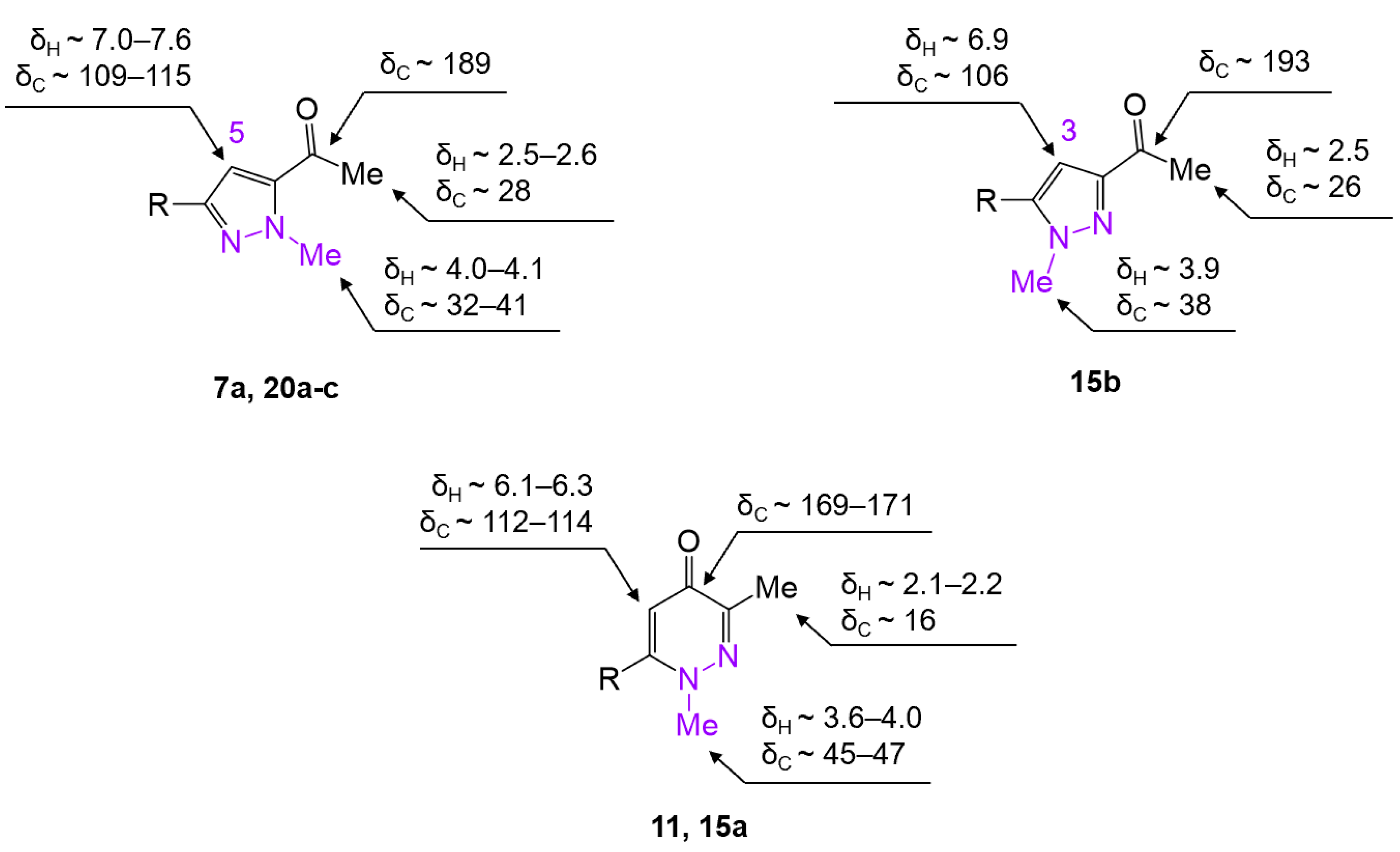
2. Results and Discussion
3. Experimental
3.1. Materials and Methods
3.2. General Procedures
3.3. Spectral and Elemental Analysis Data of Synthesized Compounds
3.4. XRD Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ebenezer, O.; Shapi, M.; Tuszynski, J.A. A Review of the recent development in the synthesis and biological evaluations of pyrazole derivatives. Biomedicines 2022, 10, 1124. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-aizari, F.A.; Ansar, M. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; Dos Santos, M.S.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef]
- Khan, M.F.; Alam, M.M.; Verma, G.; Akhtar, W.; Akhter, M.; Shaquiquzzaman, M. The therapeutic voyage of pyrazole and its analogs: A review. Eur. J. Med. Chem. 2016, 120, 170–201. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman, S. Review: Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Verma, R.; Verma, S.K.; Rakesh, K.P.; Girish, Y.R.; Ashrafizadeh, M.; Sharath Kumar, K.S.; Rangappa, K.S. Pyrazole-based analogs as potential antibacterial agents against methicillin-resistance staphylococcus aureus (MRSA) and its SAR elucidation. Eur. J. Med. Chem. 2021, 212, 113134. [Google Scholar] [CrossRef] [PubMed]
- Bennani, F.E.; Doudach, L.; Cherrah, Y.; Ramli, Y.; Karrouchi, K.; Ansar, M.; Faouzi, M.E.A. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 2020, 97, 103470. [Google Scholar] [CrossRef]
- Brullo, C.; Rapetti, F.; Bruno, O. Pyrazolyl-Ureas as Interesting Scaffold in Medicinal Chemistry. Molecules 2020, 25, 3457. [Google Scholar] [CrossRef]
- Alam, M.J.; Alam, O.; Naim, M.J.; Nawaz, F.; Manaithiya, A.; Imran, M.; Thabet, H.K.; Alshehri, S.; Ghoneim, M.M.; Alam, P.; et al. Recent Advancement in Drug Design and Discovery of Pyrazole Biomolecules as Cancer and Inflammation Therapeutics. Molecules 2022, 27, 8708. [Google Scholar] [CrossRef]
- Asif, M. Some Recent Approaches of Biologically Active Substituted Pyridazine and Phthalazine Drugs. Curr. Med. Chem. 2012, 19, 2984–2991. [Google Scholar] [CrossRef]
- Imran, M.; Asif, M. Biologically Active Pyridazines and Pyridazinone Derivatives: A Scaffold for the Highly Functionalized Compounds. Russ. J. Bioorg. Chem. 2020, 46, 726–744. [Google Scholar] [CrossRef]
- He, Z.-X.; Gong, Y.-P.; Zhang, X.; Ma, L.-Y.; Zhao, W. Pyridazine as a privileged structure: An updated review on anticancer activity of pyridazine containing bioactive molecules. Eur. J. Med. Chem. 2020, 209, 112946. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G. Are pyridazines privileged structures? Med. Chem. Commun. 2011, 2, 935. [Google Scholar] [CrossRef]
- Abida, M.; Alam, T.; Asif, M. Pharmacological activities of pyridazines and pyridazinone Derivatives: A Review on biologically active scaffold. South Asian Res. J. Pharm. Sci. 2019, 1, 16–37. [Google Scholar]
- Meanwell, N.A. The pyridazine heterocycle in molecular recognition and drug discovery. Med. Chem. Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.F.; Turones, L.C.; Cavalcante, K.V.N.; Rosa Júnior, I.A.; Xavier, C.H.; Rosseto, L.P.; Napolitano, H.B.; Castro, P.F.dS.; Neto, M.L.F.; Galvão, G.M.; et al. Heterocyclic Compounds: Pharmacology of Pyrazole Analogs from Rational Structural Considerations. Front. Pharmacol. 2021, 12, 666725. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Zaraei, S.-O.; Madkour, M.M.; Anbar, H.S. Evaluation of substituted pyrazole-based kinase inhibitors in one decade (2011–2020): Current status and future prospects. Molecules 2022, 27, 330. [Google Scholar] [CrossRef]
- Clemett, D.; Goa, K.L. Celecoxib: A review of its use in osteoarthritis, rheumatoid arthritis and acute pain. Drugs 2000, 59, 957–980. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R. Ruxolitinib: The First FDA Approved Therapy for the Treatment of Myelofibrosis. Clin. Cancer Res. 2012, 18, 3008–3014. [Google Scholar] [CrossRef]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef]
- Menichincheri, M.; Ardini, E.; Magnaghi, P.; Avanzi, N.; Banfi, P.; Bossi, R.; Buffa, L.; Canevari, G.; Ceriani, L.; Colombo, M.; et al. Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor. J. Med. Chem. 2016, 59, 3392–3408. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Cheremina, O.; Bachmakov, J.; Renner, B.; Zolk, O.; Fromm, M.F.; Brune, K. Dipyrone elicits substantial inhibition of peripheral cyclooxygenases in humans: New insights into the pharmacology of an old analgesic. FASEB J. 2007, 21, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Dooley, M.; Plosker, G.L. Zaleplon: A review of its use in the treatment of insomnia. Drugs 2000, 60, 413–445. [Google Scholar] [CrossRef] [PubMed]
- Papp, Z.; Édes, I.; Fruhwald, S.; De Hert, S.G.; Salmenperä, M.; Leppikangas, H.; Mebazaa, A.; Landoni, G.; Grossini, E.; Caimmi, P.; et al. Levosimendan: Molecular mechanisms and clinical implications. Int. J. Cardiol. 2012, 159, 82–87. [Google Scholar] [CrossRef]
- Bernstein, J.A. Azelastine hydrochloride: A review of pharmacology, pharmacokinetics, clinical efficacy and tolerability. Curr. Med. Res. Opin. 2007, 23, 2441–2452. [Google Scholar] [CrossRef]
- Deeks, E.D. Olaparib: First Global Approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef]
- Contreras, J.-M.; Rival, Y.M.; Chayer, S.; Bourguignon, J.-J.; Wermuth, C.G. Aminopyridazines as Acetylcholinesterase Inhibitors. J. Med. Chem. 1999, 42, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, W.; Shaquiquzzaman, M.; Akhter, M.; Verma, G.; Khan, M.F.; Alam, M.M. The therapeutic journey of pyridazinone. Eur. J. Med. Chem. 2016, 123, 256–281. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Bhosle, P.A. Pyridazinone: An important element of pharmacophore possessing broad spectrum of activity. Med. Chem. Res. 2015, 24, 3579–3598. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, D.; Bansal, R. Pyridazinone: An attractive lead for anti-inflammatory and analgesic drug discovery. Future Med. Chem. 2017, 9, 95–127. [Google Scholar] [CrossRef]
- Singh, J.; Kumar, v.; Silakari, P.; Kumar, S. Pyridazinones: A versatile scaffold in the development of potential target-based novel anticancer agents. J. Heterocycl. Chem. 2022, 60, 929–949. [Google Scholar] [CrossRef]
- Daoui, S.; Direkel, Ş.; Ibrahim, M.M.; Tüzün, B.; Chelfi, T.; Al-Ghorbani, M.; Bouatia, M.; Karbane, M.E.; Doukkali, A.; Benchat, N.; et al. Synthesis, Spectroscopic Characterization, Antibacterial Activity, and Computational Studies of Novel Pyridazinone Derivatives. Molecules 2023, 28, 678. [Google Scholar] [CrossRef]
- Hassan, M.S.A.; Ahmed, E.M.; El-Malah, A.A.; Kassab, A.E. Anti-inflammatory activity of pyridazinones: A review. Arch. Pharm. 2022, 355, 2200067. [Google Scholar] [CrossRef] [PubMed]
- Lamberth, C. Pyrazole Chemistry in Crop Protection. Heterocycles 2007, 71, 1467–1502. [Google Scholar] [CrossRef]
- Lamberth, C. Pyridazine Chemistry in Crop Protection. J. Heterocycl. Chem. 2017, 54, 2974–2984. [Google Scholar] [CrossRef]
- Chalifour, A.; Arts, M.T.; Kainz, M.J.; Juneau, P. Combined effect of temperature and bleaching herbicides on photosynthesis, pigment and fatty acid composition of Chlamydomonas reinhardtii. Eur. J. Phycol. 2014, 49, 508–515. [Google Scholar] [CrossRef]
- Fernández-Pérez, M.; Villafranca-Sánchez, M.; Flores-Céspedes, F.; Daza-Fernández, I. Ethylcellulose and lignin as bearer polymers in controlled release formulations of chloridazon. Carbohydr. Polym. 2011, 83, 1672–1679. [Google Scholar] [CrossRef]
- Liu, C.; Lu, D.; Wang, Y.; Huang, J.; Wan, K.; Wang, F. Residue and risk assessment of pyridaben in cabbage. Food Chem. 2014, 149, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Vidau, C.; Brunet, J.-L.; Badiou, A.; Belzunces, L.P. Phenylpyrazole insecticides induce cytotoxicity by altering mechanisms involved in cellular energy supply in the human epithelial cell model Caco-2. Toxicol. Vitr. 2009, 23, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Simon-Delso, N.; Amaral-Rogers, V.; Belzunces, L.P.; Bonmatin, J.M.; Chagnon, M.; Downs, C.; Furlan, L.; Gibbons, D.W.; Giorio, C.; Girolami, V.; et al. Systemic insecticides (neonicotinoids and fipronil): Trends, uses, mode of action and metabolites. Environ. Sci. Pollut. Res. 2014, 22, 5–34. [Google Scholar] [CrossRef] [PubMed]
- Khalighi, M.; Dermauw, W.; Wybouw, N.; Bajda, S.; Osakabe, M.; Tirry, L.; Van Leeuwen, T. Molecular analysis of cyenopyrafen resistance in the two-spotted spider mite Tetranychus urticae. Pest Manag. Sci. 2015, 72, 103–112. [Google Scholar] [CrossRef]
- Dekeyser, M.A. Acaricide mode of action. Pest Manag. Sci. 2005, 61, 103–110. [Google Scholar] [CrossRef]
- Mykhailiuk, P.K. Fluorinated pyrazoles: From synthesis to applications. Chem. Rev. 2021, 121, 1670–1715. [Google Scholar] [CrossRef]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Singh, S.P.; Kumar, V.; Aggarwal, R. Reaction of Hydrazines and Hydroxylamine with Trifluoromethyl-β-diketones: Synthesis of Trifluoromethylpyrazole and Isoxazole Derivatives. Heterocycles 2008, 75, 2893–2929. [Google Scholar] [CrossRef]
- Wang, H.; Sun, X.; Zhang, S.; Liu, G.; Wang, C.; Zhu, L.; Zhang, H. Efficient Copper-Catalyzed Synthesis of Substituted Pyrazoles at Room Temperature. Synlett 2018, 29, 2689–2692. [Google Scholar] [CrossRef]
- Rulev, A.Y.; Romanov, A.R. Unsaturated polyfluoroalkyl ketones in the synthesis of nitrogen-bearing heterocycles. RSC Adv. 2016, 6, 1984–1998. [Google Scholar] [CrossRef]
- Zhang, X.; Kang, J.; Niu, P.; Wu, J.; Yu, W.; Chang, J. I2-Mediated Oxidative C–N Bond Formation for Metal-Free One-Pot Synthesis of Di-, Tri-, and Tetrasubstituted Pyrazoles from α,β-Unsaturated Aldehydes/Ketones and Hydrazines. J. Org. Chem. 2014, 79, 10170–10178. [Google Scholar] [CrossRef]
- Baiju, T.V.; Namboothiri, I.N.N. Synthesis of Functionalized Pyrazoles via 1,3-Dipolar Cycloaddition of α-Diazo-β-ketophosphonates, Sufones and Esters with Electron-Deficient Alkenes. Chem. Rec. 2017, 17, 939–955. [Google Scholar] [CrossRef]
- Chandrasekharan, S.P.; Dhami, A.; Kumara, S.; Mohanan, K. Recent advances in pyrazole synthesis employing diazo compounds and synthetic analogues. Org. Biomol. Chem. 2022, 20, 8787–8817. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Nie, J.; Sun, L.; Zheng, Y.; Ma, J.-A. Silver-Mediated Cycloaddition of Alkynes with CF3CHN2: Highly Regioselective Synthesis of 3-Trifluoromethylpyrazoles. Angew. Chem. Int. Ed. 2013, 52, 6255–6258. [Google Scholar] [CrossRef] [PubMed]
- Kula, K.; Łapczuk, A.; Sadowski, M.; Kras, J.; Zawadzińska, K.; Demchuk, O.M.; Gaurav, G.K.; Wróblewska, A.; Jasiński, R. On the Question of the Formation of Nitro-Functionalized 2,4-Pyrazole Analogs on the Basis of Nitrylimine Molecular Systems and 3,3,3-Trichloro-1-Nitroprop-1-Ene. Molecules 2022, 27, 8409. [Google Scholar] [CrossRef] [PubMed]
- Mykhailiuk, P.K. In Situ Generation of Difluoromethyl Diazomethane for [3+2] Cycloadditions with Alkynes. Angew. Chem. Int. Ed. 2015, 54, 6558–6561. [Google Scholar] [CrossRef]
- Giovannoni, M.P.; Schepetkin, I.A.; Cilibrizzi, A.; Crocetti, L.; Khlebnikov, A.I.; Dahlgren, C.; Graziano, A.; Dal Piaz, V.; Kirpotina, L.N.; Zerbinati, S.; et al. Further studies on 2-arylacetamide pyridazin-3(2H)-ones: Design, synthesis and evaluation of 4,6-disubstituted analogs as formyl peptide receptors (FPRs) agonists. Eur. J. Med. Chem. 2013, 64, 512–528. [Google Scholar] [CrossRef] [PubMed]
- Abdelbaset, M.S.; Abuo-Rahma, G.E.-D.A.; Abdelrahman, M.H.; Ramadan, M.; Youssif, B.G.M.; Bukhari, S.N.A.; Mohamed, M.F.A.; Abdel-Aziz, M. Novel pyrrol-2(3H)-ones and pyridazin-3(2H)-ones carrying quinoline scaffold as anti-proliferative tubulin polymerization inhibitors. Bioorg. Chem. 2018, 80, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Barberot, C.; Moniot, A.; Allart-Simon, I.; Malleret, L.; Yegorova, T.; Laronze-Cochard, M.; Bentaher, A.; Médebielle, M.; Bouillon, J.-P.; Hénon, E.; et al. Synthesis and biological evaluation of pyridazinone derivatives as potential anti-inflammatory agents. Eur. J. Med. Chem. 2018, 146, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.Y.; Aly, A.F.; Abdullah, N.H.; Ismail, M.F. Synthesis, Characterization and Antifungal Evaluation of Novel Pyridazin-3(2H)-One Derivatives. Polycycl. Aromat. Compd. 2023, 43, 2356–2375. [Google Scholar] [CrossRef]
- Prime, M.E.; Courtney, S.M.; Brookfield, F.A.; Marston, R.W.; Walker, V.; Warne, J.; Boyd, A.E.; Kairies, N.A.; von der Saal, W.; Limberg, A.; et al. Phthalazinone Pyrazoles as Potent, Selective, and Orally Bioavailable Inhibitors of Aurora-A Kinase. J. Med. Chem. 2011, 54, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Elagawany, M.; Ibrahim, M.A.; Ali Ahmed, H.E.; El-Etrawy, A.S.; Ghiaty, A.; Abdel-Samii, Z.K.; El-Feky, S.A.; Bajorath, J. Design, synthesis, and molecular modelling of pyridazinone and phthalazinone derivatives as protein kinases inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Shao, P.-L.; He, Y. Synthesis of 1,4,5,6-tetrahydropyridazines and pyridazines via transition-metal-free (4 + 2) cycloaddition of alkoxyallenes with 1,2-diaza-1,3-dienes. RSC Adv. 2019, 9, 21507–21512. [Google Scholar] [CrossRef] [PubMed]
- Schnell, S.D.; González, J.A.; Sklyaruk, J.; Linden, A.; Gademann, K. Boron Trifluoride-Mediated Cycloaddition of 3-Bromotetrazine and Silyl Enol Ethers: Synthesis of 3-Bromo-pyridazines. J. Org. Chem. 2021, 86, 12008–12023. [Google Scholar] [CrossRef] [PubMed]
- Obydennov, D.L.; Khammatova, L.R.; Eltsov, O.S.; Sosnovskikh, V.Y. A chemo- and regiocontrolled approach to bipyrazoles and pyridones via the reaction of ethyl 5-acyl-4-pyrone-2-carboxylates with hydrazines. Org. Biomol. Chem. 2018, 16, 1692–1707. [Google Scholar] [CrossRef]
- Fedin, V.V.; Usachev, S.A.; Obydennov, D.L.; Sosnovskikh, V.Y. Reactions of Trifluorotriacetic Acid Lactone and Hexafluorodehydroacetic Acid with Amines: Synthesis of Trifluoromethylated 4-Pyridones and Aminoenones. Molecules 2022, 27, 7098. [Google Scholar] [CrossRef]
- Fandrick, D.R.; Sanyal, S.; Kaloko, J.; Mulder, J.A.; Wang, Y.; Wu, L.; Lee, H.; Roschangar, F.; Hoffmann, M.; Senanayake, C.H. A Michael Equilibration Model to Control Site Selectivity in the Condensation toward Aminopyrazoles. Org. Lett. 2015, 17, 2964–2967. [Google Scholar] [CrossRef]
- Shaitanova, E.N.; Balabon, O.A.; Rybakova, A.N.; Khlebnicova, T.S.; Lakhvich, F.A.; Gerus, I.I. Synthesis of functionalized fluoroalkyl pyrimidines and pyrazoles from fluoroalkyl enones. J. Fluor. Chem. 2021, 252, 109905. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Ivanova, O.A.; Shumsky, A.N.; Trushkov, I.V. Synthesis of hexahydropyridazin-3-ones by reactions between donor-acceptor cyclopropanes and phenylhydrazine. Chem. Heterocycl. Compd. 2017, 53, 1220–1227. [Google Scholar] [CrossRef]
- Shokova, E.A.; Kim, J.K.; Kovalev, V.V. 1,3-Diketones. Synthesis and properties. Russ. J. Org. Chem. 2015, 51, 755–830. [Google Scholar] [CrossRef]
- Atta, K.F.M.; Farahat, O.O.M.; Al-Shargabi, T.Q.; Marei, M.G.; El Ashry, E.S.H. Chemistry of Pent-4-yne-1,3-diones (Acetylenic β-diketones) as Precursors for Heterocyclic Compounds. Adv. Heterocycl. Chem. 2014, 113, 67–110. [Google Scholar] [CrossRef]
- Abdelhamid, A.O.; Gomha, S.M. The Chemistry of acetylpyrazoles and its utility in heterocyclic synthesis. J. Heterocycl. Chem. 2019, 56, 726–758. [Google Scholar] [CrossRef]
- Joksimović, N.; Janković, N.; Davidović, G.; Bugarčić, Z. 2,4-Diketo esters: Crucial intermediates for drug discovery. Bioorg. Chem. 2020, 105, 104343. [Google Scholar] [CrossRef] [PubMed]
- Dawood, K.M.; Abdel-Gawad, H.; Mohamed, H.A.; Abdel-Wahab, B.F. Utility of 2,4-dioxoesters in the synthesis of new heterocycles. Heterocycles 2010, 81, 1–55. [Google Scholar] [CrossRef]
- Bazhin, D.N.; Kudyakova, Y.S.; Burgart, Y.V.; Saloutin, V.I. Intramolecular cyclization of lithium 4,4-dimethoxy-1-(perfluoroalkyl)pentane-1,3-dionates on treatment with boron trifluoride diethyl etherate. Russ. Chem. Bull. 2018, 67, 497–499. [Google Scholar] [CrossRef]
- Kudyakova, Y.S.; Onoprienko, A.Y.; Edilova, Y.O.; Burgart, Y.V.; Saloutin, V.I.; Bazhin, D.N. Effect of the nature of a fluorinated substituent on the synthesis of functionalized 1,3-diketones. Russ. Chem. Bull. 2021, 70, 745–752. [Google Scholar] [CrossRef]
- Chizhov, D.L.; Belyaev, D.V.; Yachevskii, D.S.; Rusinov, G.L.; Chupakhin, O.N.; Charushin, V.N. Efficient and scalable synthesis of 3-(polyfluoroacyl)pyruvaldehydes dimethyl acetals: A novel functionalized fluorinated building-block. J. Fluor. Chem. 2017, 199, 39–45. [Google Scholar] [CrossRef]
- Safrygin, A.V.; Irgashev, R.A.; Slepukhin, P.A.; Röschenthaler, G.-V.; Sosnovskikh, V.Y. Synthesis of 5-aryl-2-hydroxy-2-(trifluoromethyl)furan-3(2H)-ones and their reactions with aromatic 1,2-diamines, hydrazine and hydroxylamine. Tetrahedron 2015, 71, 8535–8543. [Google Scholar] [CrossRef]
- Irgashev, R.A.; Safrygin, A.V.; Ezhikova, M.A.; Kodess, M.I.; Röschenthaler, G.-V.; Sosnovskikh, V.Y. Synthesis of 2-(trifluoroacetyl)chromones and their reactions with 1,2-diamines. Tetrahedron 2015, 71, 1822–1830. [Google Scholar] [CrossRef]
- Bazhin, D.N.; Kudyakova, Y.S.; Edilova, Y.O.; Burgart, Y.V.; Saloutin, V.I. Fluorinated 1,2,4-triketone analogs: New prospects for heterocyclic and coordination chemistry. Russ. Chem. Bull. 2022, 71, 1321–1341. [Google Scholar] [CrossRef]
- Edilova, Y.O.; Kudyakova, Y.S.; Kiskin, M.A.; Burgart, Y.V.; Saloutin, V.I.; Bazhin, D.N. Expanding 1,2,4-triketone toolbox for use as fluorinated building blocks in the synthesis of pyrazoles, pyridazinones and β-diketohydrazones. J. Fluor. Chem. 2022, 253, 109932. [Google Scholar] [CrossRef]
- Bazhin, D.N.; Kudyakova, Y.S.; Röschenthaler, G.-V.; Burgart, Y.V.; Slepukhin, P.A.; Isenov, M.L.; Saloutin, V.I.; Charushin, V.N. A convenient approach to CF3-containing N-heterocycles based on 2-methoxy-2-methyl-5-(trifluoromethyl)furan-3(2H)-one. Eur. J. Org. Chem. 2015, 23, 5236–5245. [Google Scholar] [CrossRef]
- Kudyakova, Y.S.; Onoprienko, A.Y.; Slepukhin, P.A.; Burgart, Y.V.; Saloutin, V.I.; Bazhin, D.N. Fluorine-containing furan-3(2H)-ones in reactions with binucleophiles: CF3 vs C2F5. Chem. Heterocycl. Compd. 2019, 55, 517–522. [Google Scholar] [CrossRef]
- Bazhin, D.N.; Chizhov, D.L.; Röschenthaler, G.-V.; Kudyakova, Y.S.; Burgart, Y.V.; Slepukhin, P.A.; Saloutin, V.I.; Charushin, V.N. A concise approach to CF3-containing furan-3-ones, (bis)pyrazoles from novel fluorinated building blocks based on 2,3-butanedione. Tetrahedron Lett. 2014, 55, 5714–5717. [Google Scholar] [CrossRef]
- Berens, U.; Leckel, D.; Oepen, S.C. Transacetalization of diethyl tartrate with acetals of α-dicarbonyl compounds: A simple access to a new class of C2-symmetric auxiliaries and ligands. J. Org. Chem. 1995, 60, 8204–8208. [Google Scholar] [CrossRef]
- Tsubusaki, T.; Nishino, H. Formation of 1,2-Dioxolanes Using Mn(III)-Based Reaction of Various Arylacetylenes with 2,4-Pentanedione and Related Reaction. Tetrahedron 2009, 65, 3745–3752. [Google Scholar] [CrossRef]
- Zbiral, E.; Bauer, E. Reaktionen mit phosphororganischen verbindungen—XXXII: Zur umsetzung von β-acylvinylphosphoniumsalzen mit diazoverbindungen. Tetrahedron 1972, 28, 4189–4196. [Google Scholar] [CrossRef]
- Rateb, L.; Soliman, G. 286. Synthesis of Heterocyclic Compounds from δ-Unsaturated 1,3-Diketo-Esters. Part II. α-Substituted Styrylpyrazole- and Styrylisoxazole-Carboxylic Esters. J. Chem. Soc. 1960, 1426–1430. [Google Scholar] [CrossRef]
- SMART (Control) and SAINT (Integration) Software, Version 5.0; Bruker AXS, Inc.: Madison, WI, USA, 1997.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2007, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Conditions | Products (Ratio) | Products (Yields) |
|---|---|---|---|
| 2b | i | 13a:13b (4:1) | 13a (59%) |
| 2b | ii | 14 | 14 (83%) |
| 2b | iii | 15a:15b (3:1) | 15a (61%), 15b (24%) |
| 2b | iv | 15a:15b (1:3) | – |
| 4a | iii | 15a:15b (3:1) | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edilova, Y.O.; Osipova, E.A.; Slepukhin, P.A.; Saloutin, V.I.; Bazhin, D.N. Exploring Three Avenues: Chemo- and Regioselective Transformations of 1,2,4-Triketone Analogs into Pyrazoles and Pyridazinones. Int. J. Mol. Sci. 2023, 24, 14234. https://doi.org/10.3390/ijms241814234
Edilova YO, Osipova EA, Slepukhin PA, Saloutin VI, Bazhin DN. Exploring Three Avenues: Chemo- and Regioselective Transformations of 1,2,4-Triketone Analogs into Pyrazoles and Pyridazinones. International Journal of Molecular Sciences. 2023; 24(18):14234. https://doi.org/10.3390/ijms241814234
Chicago/Turabian StyleEdilova, Yulia O., Ekaterina A. Osipova, Pavel A. Slepukhin, Victor I. Saloutin, and Denis N. Bazhin. 2023. "Exploring Three Avenues: Chemo- and Regioselective Transformations of 1,2,4-Triketone Analogs into Pyrazoles and Pyridazinones" International Journal of Molecular Sciences 24, no. 18: 14234. https://doi.org/10.3390/ijms241814234