
Synergistic Pulmonoprotective Effect of Natural Prolyl Oligopeptidase Inhibitors in In Vitro and In Vivo Models of Acute Respiratory Distress Syndrome
 , , and
, , and
Abstract
:1. Introduction
2. Results
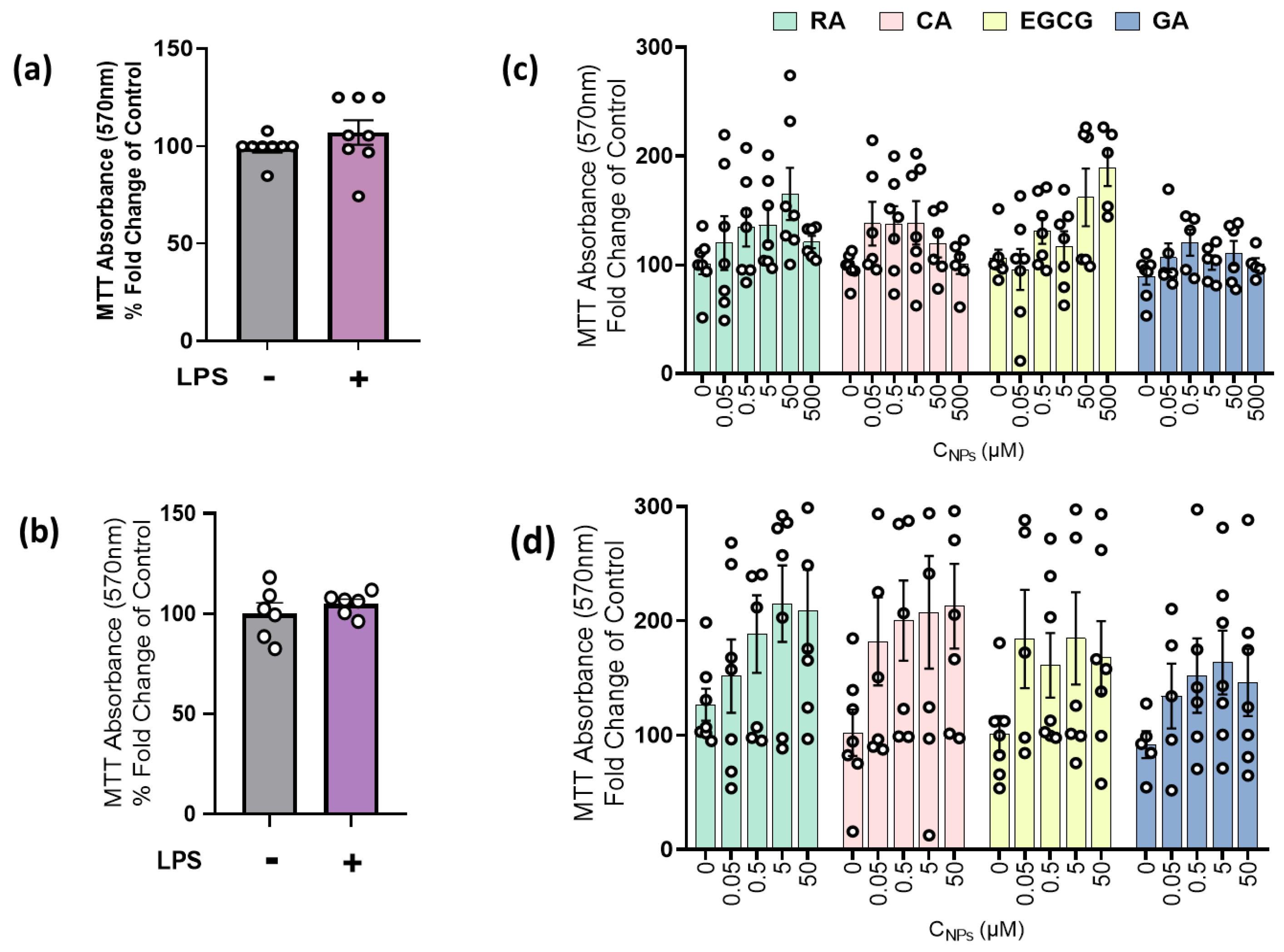
2.1. LPS and NPs Did Not Exert Cytotoxicity in RAW264.7 Cells and MLECs
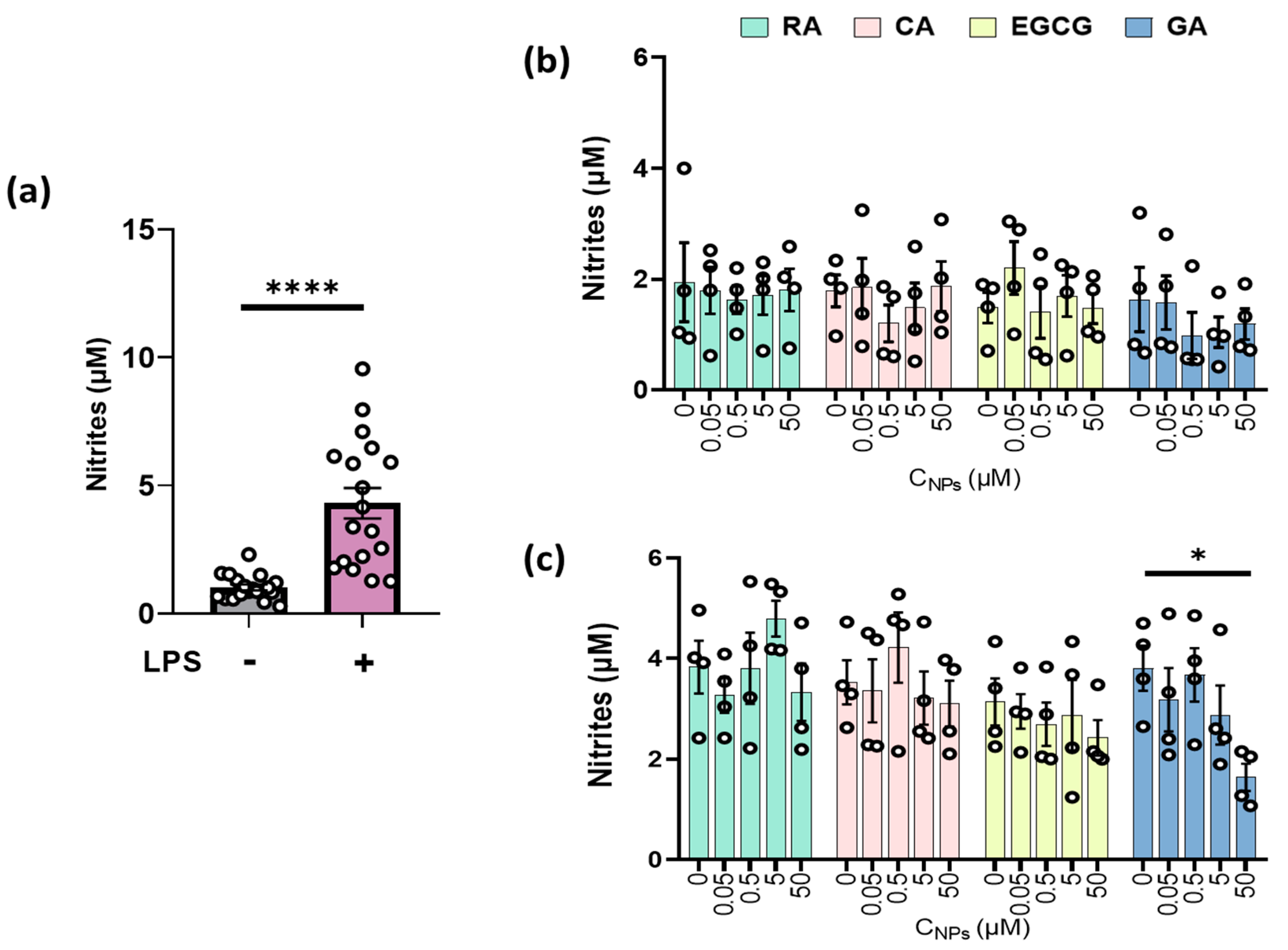
2.2. GA Reduced Nitrite Production in LPS-Stimulated RAW264.7 Cells
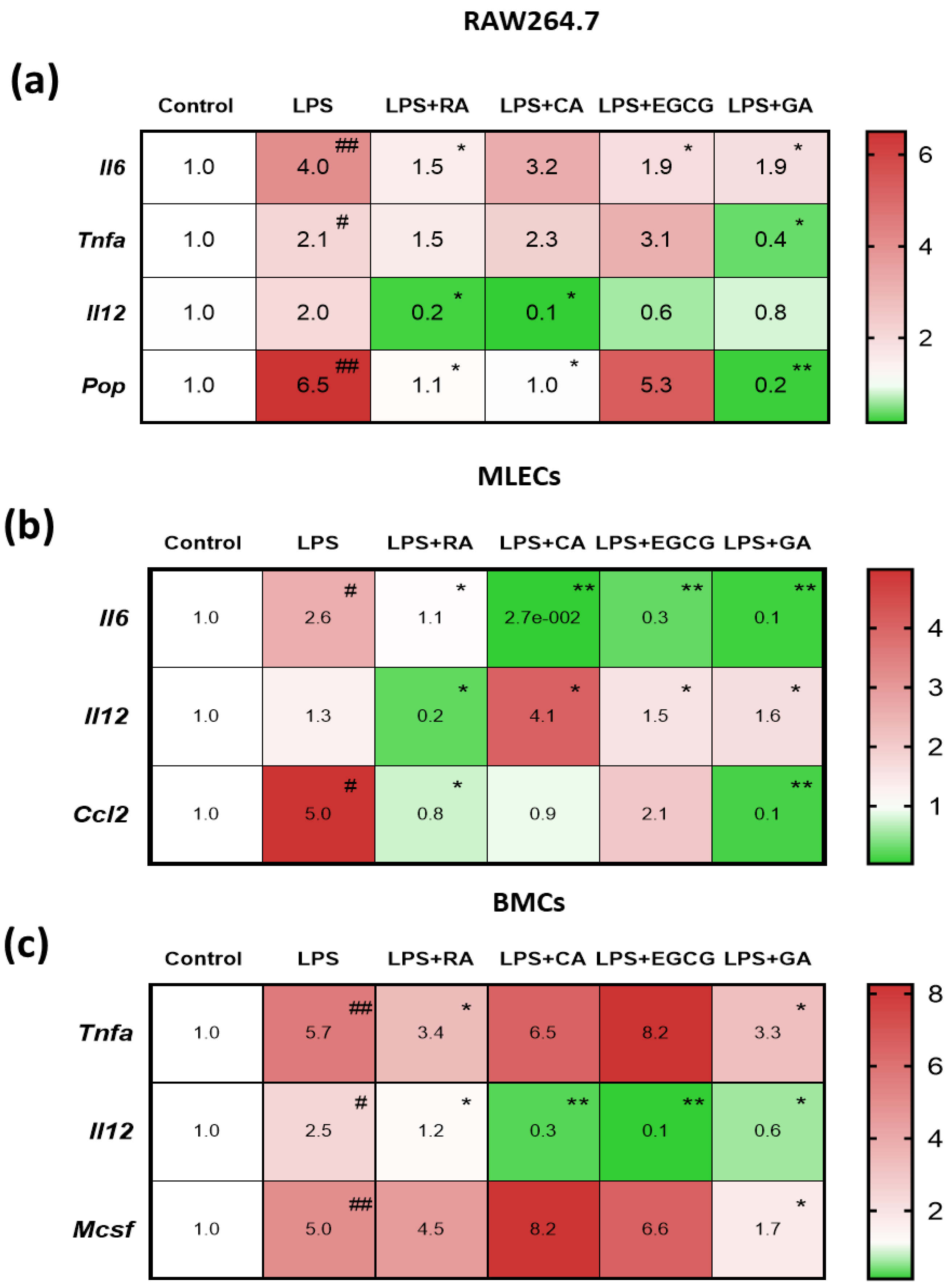
2.3. NPs Suppressed the LPS-Elevated Expression of Pro-Inflammatory Mediators in RAW264.7 Macrophages and in Primary Lung Epithelial and Bone Marrow Cells
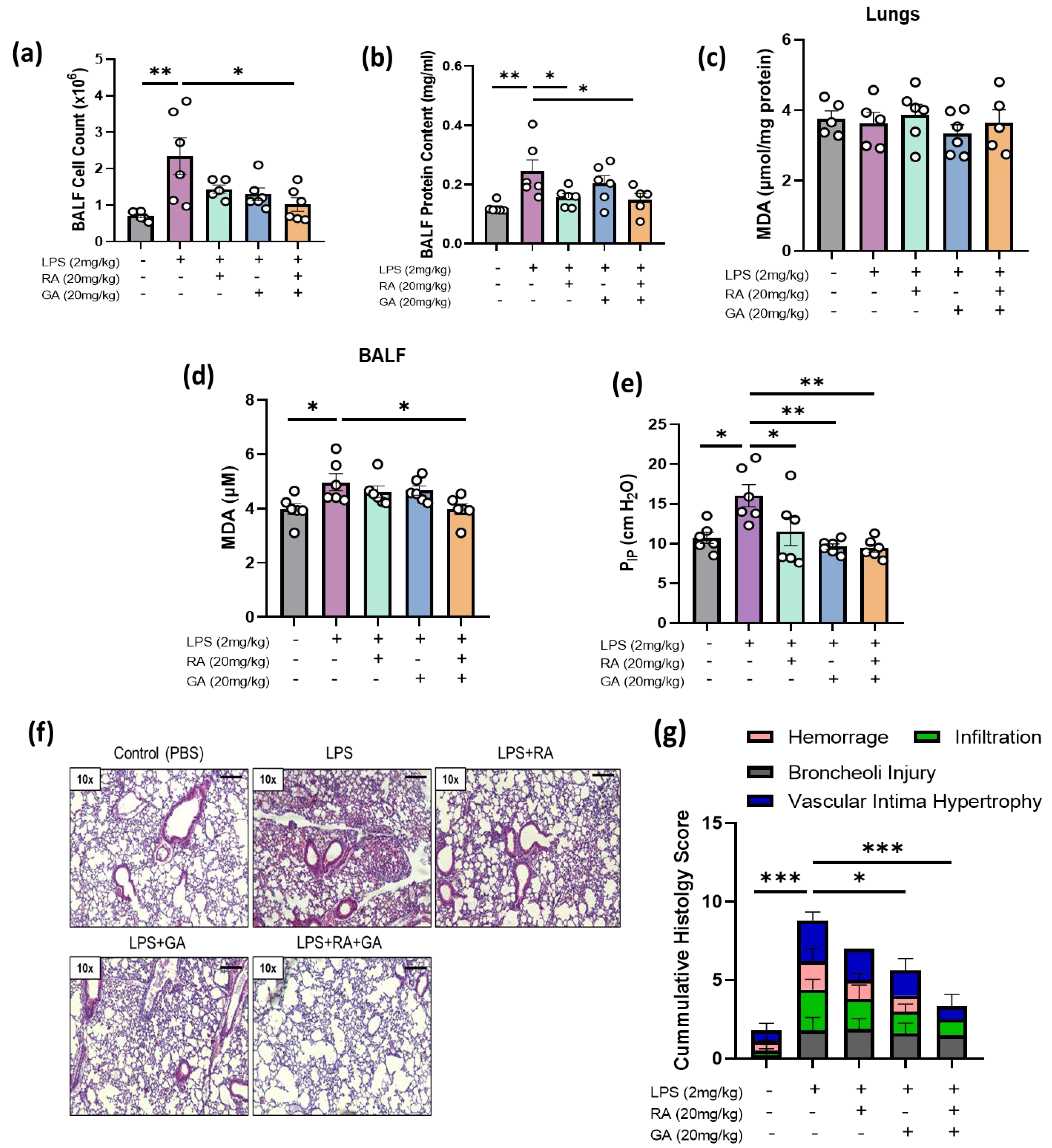
2.4. RA, GA, and Their Co-administration Alleviated LPS-Induced ARDS In Vivo
2.5. Co-Administration of RA and GA Decreased LPS-Elevated Lipid Peroxidation Levels in BALF
2.6. RA, GA, and Their Co-Administration Ameliorated Respiratory Capacity of LPS-Challenged Mice
2.7. RA, GA, and Mainly Their Co-Administration Alleviated LPS-Induced Histologic Changes in Lung Tissues
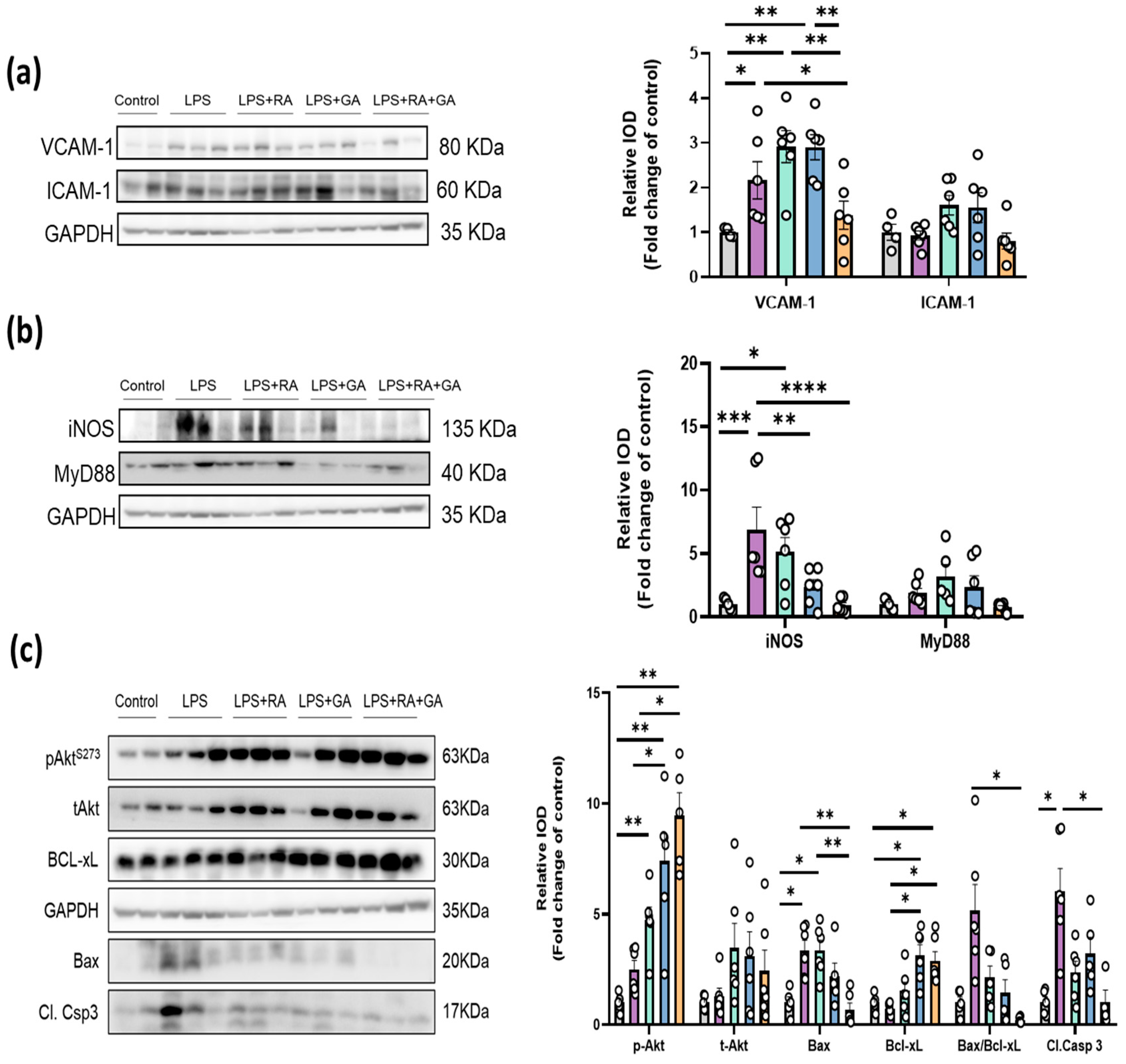
2.8. RA, GA and Their Co-Administration Suppressed the Activation of Key LPS-Induced Inflammation and Apoptotic Mechanisms
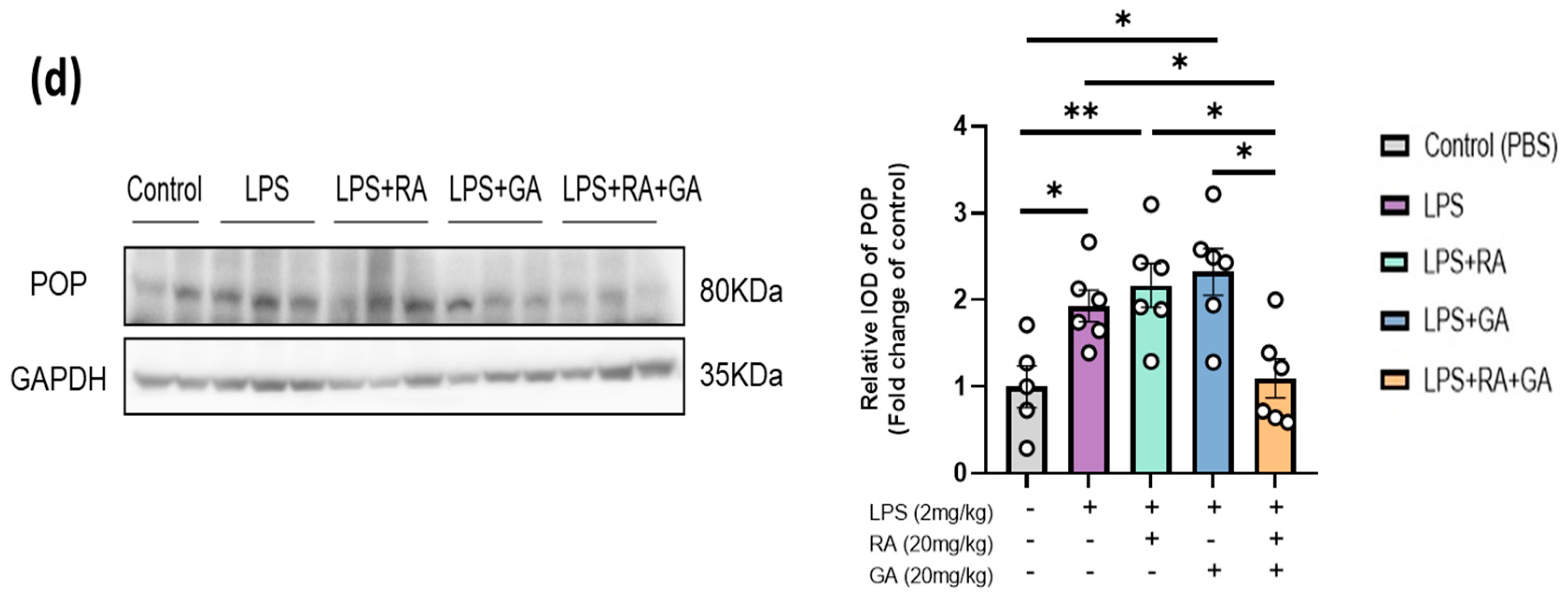
2.9. RA and GA Co-Administration Reduced LPS-Elevated POP Expression
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Isolation of Primary Mouse Lung Epithelial Cells (MLECs)
4.3. Isolation of Primary Mouse Bone Marrow Cells (BMCs)
4.4. Cell Cultures
4.5. Evaluation of Cell Viability
4.6. Assessment of NO Production of RAW264.7 Macrophages
4.7. RNA Extraction, cDNA Synthesis and RT-PCR
4.8. Animals
4.9. In Vivo Experimental Protocol of LPS-Induced ARDS Model
4.10. BALF Collection and Inspiratory Capacity Determination
4.11. Histopathologic Evaluation
4.12. Determination of MDA Levels in BALF and Lung Tissues
4.13. Western Blot Analysis
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Dushianthan, A.; Grocott, M.P.; Postle, A.D.; Cusack, R. Acute respiratory distress syndrome and acute lung injury. Postgrad. Med. J. 2011, 87, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef]
- Hasan, S.S.; Capstick, T.; Ahmed, R.; Kow, C.S.; Mazhar, F.; Merchant, H.A.; Zaidi, S.T.R. Mortality in COVID-19 patients with acute respiratory distress syndrome and corticosteroids use: A systematic review and meta-analysis. Expert Rev. Respir. Med. 2020, 14, 1149–1163. [Google Scholar] [CrossRef]
- Ragaller, M.; Richter, T. Acute lung injury and acute respiratory distress syndrome. J. Emergencies Trauma Shock 2010, 3, 43–51. [Google Scholar] [CrossRef]
- Rehberg, S.; Ertmer, C.; Westphal, M. Mechanical ventilation in patients with ARDS: Is the lung’s fortune the right ventricle’s poison? Intensive Care Med. 2009, 35, 1825–1826. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Chinn, A.J.; Leeper, K.V.; Wunderink, R.G.; Tolley, E.; Winer-Muram, H.T.; Khare, V.; Eltorky, M. Corticosteroid rescue treatment of progressive fibroproliferation in late ARDS. Patterns of response and predictors of outcome. Chest 1994, 105, 1516–1527. [Google Scholar] [CrossRef]
- Bos, L.D.J.; Ware, L.B. Acute respiratory distress syndrome: Causes, pathophysiology, and phenotypes. Lancet 2022, 400, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Cardinal-Fernandez, P.; Lorente, J.A.; Ballen-Barragan, A.; Matute-Bello, G. Acute Respiratory Distress Syndrome and Diffuse Alveolar Damage. New Insights on a Complex Relationship. Ann. Am. Thorac. Soc. 2017, 14, 844–850. [Google Scholar] [CrossRef]
- Bellingan, G.J. The pulmonary physician in critical care * 6: The pathogenesis of ALI/ARDS. Thorax 2002, 57, 540–546. [Google Scholar] [CrossRef]
- Crimi, E.; Slutsky, A.S. Inflammation and the acute respiratory distress syndrome. Best Pract. Res. Clin. Anaesthesiol. 2004, 18, 477–492. [Google Scholar] [CrossRef]
- Puneet, P.; Moochhala, S.; Bhatia, M. Chemokines in acute respiratory distress syndrome. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L3–L15. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Q.; Zhou, C.C.; Yu, L.Y.; Wang, L.; Deng, J.L.; Tao, Y.L.; Zhang, F.; Chen, W.S. Natural product derived phytochemicals in managing acute lung injury by multiple mechanisms. Pharmacol. Res. 2021, 163, 105224. [Google Scholar] [CrossRef] [PubMed]
- Casili, G.; Scuderi, S.A.; Lanza, M.; Filippone, A.; Basilotta, R.; Mannino, D.; Campolo, M.; Esposito, E.; Paterniti, I. The protective role of prolyl oligopeptidase (POP) inhibition in acute lung injury induced by intestinal ischemia-reperfusion. Oncotarget 2021, 12, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.F.; Snelgrove, R.J. The multifaceted roles of the matrikine Pro-Gly-Pro in pulmonary health and disease. Eur. Respir. Rev. 2018, 27, 180017. [Google Scholar] [CrossRef]
- Braber, S.; Koelink, P.J.; Henricks, P.A.; Jackson, P.L.; Nijkamp, F.P.; Garssen, J.; Kraneveld, A.D.; Blalock, J.E.; Folkerts, G. Cigarette smoke-induced lung emphysema in mice is associated with prolyl endopeptidase, an enzyme involved in collagen breakdown. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L255–L265. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, A.R.; Pyle, C.J.; Patel, D.F.; Jackson, P.L.; Hilliard, T.N.; Regamey, N.; Tan, H.L.; Brown, S.; Thursfield, R.; Short, C.; et al. Abnormal pro-gly-pro pathway and airway neutrophilia in pediatric cystic fibrosis. J. Cyst. Fibros. 2020, 19, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Xanthopoulos, A.; Giamouzis, G.; Boudoulas, K.D.; Starling, R.C.; Skoularigis, J.; Boudoulas, H.; Iliodromitis, E. ACE2, the Counter-Regulatory Renin-Angiotensin System Axis and COVID-19 Severity. J. Clin. Med. 2021, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Ci, X.; He, J.; Jiang, L.; Wei, M.; Cao, Q.; Guan, M.; Xie, X.; Deng, X.; He, J. Effects of a natural prolyl oligopeptidase inhibitor, rosmarinic acid, on lipopolysaccharide-induced acute lung injury in mice. Molecules 2012, 17, 3586–3598. [Google Scholar] [CrossRef]
- Fan, W.; Tezuka, Y.; Ni, K.M.; Kadota, S. Prolyl endopeptidase inhibitors from the underground part of Rhodiola sachalinensis. Chem. Pharm. Bull. 2001, 49, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Alagawany, M.; Abd El-Hack, M.E.; Farag, M.R.; Gopi, M.; Karthik, K.; Malik, Y.S.; Dhama, K. Rosmarinic acid: Modes of action, medicinal values and health benefits. Anim. Health Res. Rev. 2017, 18, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Sun, Q.; Park, Y. The Bioactive Effects of Chicoric Acid as a Functional Food Ingredient. J. Med. Food 2019, 22, 645–652. [Google Scholar] [CrossRef]
- Lopez-Lazaro, M.; Calderon-Montano, J.M.; Burgos-Moron, E.; Austin, C.A. Green tea constituents (-)-epigallocatechin-3-gallate (EGCG) and gallic acid induce topoisomerase I- and topoisomerase II-DNA complexes in cells mediated by pyrogallol-induced hydrogen peroxide. Mutagenesis 2011, 26, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Domscheit, H.; Hegeman, M.A.; Carvalho, N.; Spieth, P.M. Molecular Dynamics of Lipopolysaccharide-Induced Lung Injury in Rodents. Front. Physiol. 2020, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Li, Q.; Zhou, M. LPS-induced upregulation of the TLR4 signaling pathway inhibits osteogenic differentiation of human periodontal ligament stem cells under inflammatory conditions. Int. J. Mol. Med. 2019, 43, 2341–2351. [Google Scholar] [CrossRef]
- Yang, S.C.; Tsai, Y.F.; Pan, Y.L.; Hwang, T.L. Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed. J. 2021, 44, 439–446. [Google Scholar] [CrossRef]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field. Oxid. Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 2011, 6, 147–163. [Google Scholar] [CrossRef]
- Qi, Y.; Qian, L.; Sun, B.; Liu, L.; Wu, P.; Sun, L. Inhaled NO contributes to lung repair in piglets with acute respiratory distress syndrome via increasing circulating endothelial progenitor cells. PLoS ONE 2012, 7, e33859. [Google Scholar] [CrossRef]
- Lin, Y.L.; Lin, J.K. (-)-Epigallocatechin-3-gallate blocks the induction of nitric oxide synthase by down-regulating lipopolysaccharide-induced activity of transcription factor nuclear factor-kappaB. Mol. Pharmacol. 1997, 52, 465–472. [Google Scholar] [CrossRef]
- Sacco, R.E.; Waters, W.R.; Rudolph, K.M.; Drew, M.L. Comparative nitric oxide production by LPS-stimulated monocyte-derived macrophages from Ovis canadensis and Ovis aries. Comp. Immunol. Microbiol. Infect. Dis. 2006, 29, 1–11. [Google Scholar] [CrossRef]
- BenSaad, L.A.; Kim, K.H.; Quah, C.C.; Kim, W.R.; Shahimi, M. Anti-inflammatory potential of ellagic acid, gallic acid and punicalagin A&B isolated from Punica granatum. BMC Complement. Altern. Med. 2017, 17, 47. [Google Scholar] [CrossRef]
- Bae, I.K.; Min, H.Y.; Han, A.R.; Seo, E.K.; Lee, S.K. Suppression of lipopolysaccharide-induced expression of inducible nitric oxide synthase by brazilin in RAW 264.7 macrophage cells. Eur. J. Pharmacol. 2005, 513, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef] [PubMed]
- van der Vliet, A.; Eiserich, J.P.; Cross, C.E. Nitric oxide: A pro-inflammatory mediator in lung disease? Respir. Res. 2000, 1, 67–72. [Google Scholar] [CrossRef]
- Adhikari, N.K.; Burns, K.E.; Friedrich, J.O.; Granton, J.T.; Cook, D.J.; Meade, M.O. Effect of nitric oxide on oxygenation and mortality in acute lung injury: Systematic review and meta-analysis. BMJ 2007, 334, 779. [Google Scholar] [CrossRef]
- Cossio, I.; Lucas, D.; Hidalgo, A. Neutrophils as regulators of the hematopoietic niche. Blood 2019, 133, 2140–2148. [Google Scholar] [CrossRef]
- Christoffersson, G.; Phillipson, M. The neutrophil: One cell on many missions or many cells with different agendas? Cell Tissue Res. 2018, 371, 415–423. [Google Scholar] [CrossRef]
- Furze, R.C.; Rankin, S.M. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008, 125, 281–288. [Google Scholar] [CrossRef]
- Ware, L.B.; Herridge, M. Acute lung injury. Semin. Respir. Crit. Care Med. 2013, 34, 439–440. [Google Scholar] [CrossRef]
- Chen, X.; Tang, J.; Shuai, W.; Meng, J.; Feng, J.; Han, Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm. Res. 2020, 69, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Galani, V.; Tatsaki, E.; Bai, M.; Kitsoulis, P.; Lekka, M.; Nakos, G.; Kanavaros, P. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): An up-to-date cell-specific review. Pathol. Res. Pract. 2010, 206, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zou, L.; Sun, H.; Peng, J.; Gao, C.; Bao, L.; Ji, R.; Jin, Y.; Sun, S. A Review of the Anti-Inflammatory Effects of Rosmarinic Acid on Inflammatory Diseases. Front. Pharmacol. 2020, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Chung, S.J.; Lee, S.W.; Park, Y.B.; Lee, S.K.; Park, M.C. Gallic acid, a natural polyphenolic acid, induces apoptosis and inhibits proinflammatory gene expressions in rheumatoid arthritis fibroblast-like synoviocytes. Jt. Bone Spine 2013, 80, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Singla, E.; Puri, G.; Dharwal, V.; Naura, A.S. Gallic acid ameliorates COPD-associated exacerbation in mice. Mol. Cell. Biochem. 2021, 476, 293–302. [Google Scholar] [CrossRef]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Cortegiani, A.; Ippolito, M.; Einav, S. Rationale and evidence on the use of tocilizumab in COVID-19: A systematic review. Authors’ reply. Pulmonology 2021, 27, 87–88. [Google Scholar] [CrossRef]
- Kewan, T.; Covut, F.; Al-Jaghbeer, M.J.; Rose, L.; Gopalakrishna, K.V.; Akbik, B. Tocilizumab for treatment of patients with severe COVID-19: A retrospective cohort study. eClinicalMedicine 2020, 24, 100418. [Google Scholar] [CrossRef]
- Suter, P.M.; Suter, S.; Girardin, E.; Roux-Lombard, P.; Grau, G.E.; Dayer, J.M. High bronchoalveolar levels of tumor necrosis factor and its inhibitors, interleukin-1, interferon, and elastase, in patients with adult respiratory distress syndrome after trauma, shock, or sepsis. Am. Rev. Respir. Dis. 1992, 145, 1016–1022. [Google Scholar] [CrossRef]
- Qin, M.; Qiu, Z. Changes in TNF-alpha, IL-6, IL-10 and VEGF in rats with ARDS and the effects of dexamethasone. Exp. Ther. Med. 2019, 17, 383–387. [Google Scholar] [CrossRef]
- Yilmaz, V.; Yentur, S.P.; Saruhan-Direskeneli, G. IL-12 and IL-10 polymorphisms and their effects on cytokine production. Cytokine 2005, 30, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Ashida, N.; Arai, H.; Yamasaki, M.; Kita, T. Distinct signaling pathways for MCP-1-dependent integrin activation and chemotaxis. J. Biol. Chem. 2001, 276, 16555–16560. [Google Scholar] [CrossRef]
- Popova, A.; Kzhyshkowska, J.; Nurgazieva, D.; Goerdt, S.; Gratchev, A. Pro- and anti-inflammatory control of M-CSF-mediated macrophage differentiation. Immunobiology 2011, 216, 164–172. [Google Scholar] [CrossRef]
- Bozinovski, S.; Jones, J.; Beavitt, S.J.; Cook, A.D.; Hamilton, J.A.; Anderson, G.P. Innate immune responses to LPS in mouse lung are suppressed and reversed by neutralization of GM-CSF via repression of TLR-4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L877–L885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.K.; Zhou, Y.; Cao, J.; Liu, D.Y.; Wan, L.H. Rosmarinic acid ameliorates septic-associated mortality and lung injury in mice via GRP78/IRE1alpha/JNK pathway. J. Pharm. Pharmacol. 2021, 73, 916–921. [Google Scholar] [CrossRef]
- He, Z.; Zhu, Y.; Jiang, H. Inhibiting toll-like receptor 4 signaling ameliorates pulmonary fibrosis during acute lung injury induced by lipopolysaccharide: An experimental study. Respir. Res. 2009, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Severgnini, M.; Takahashi, S.; Rozo, L.M.; Homer, R.J.; Kuhn, C.; Jhung, J.W.; Perides, G.; Steer, M.; Hassoun, P.M.; Fanburg, B.L.; et al. Activation of the STAT pathway in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L1282–L1292. [Google Scholar] [CrossRef]
- Bhargava, M.; Viken, K.; Wang, Q.; Jagtap, P.; Bitterman, P.; Ingbar, D.; Wendt, C. Bronchoalveolar Lavage Fluid Protein Expression in Acute Respiratory Distress Syndrome Provides Insights into Pathways Activated in Subjects with Different Outcomes. Sci. Rep. 2017, 7, 7464. [Google Scholar] [CrossRef]
- Choi, K.C.; Lee, Y.H.; Jung, M.G.; Kwon, S.H.; Kim, M.J.; Jun, W.J.; Lee, J.; Lee, J.M.; Yoon, H.G. Gallic acid suppresses lipopolysaccharide-induced nuclear factor-kappaB signaling by preventing RelA acetylation in A549 lung cancer cells. Mol. Cancer Res. 2009, 7, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.; Eduardo-Figueira, M.; Barateiro, A.; Fernandes, A.; Brites, D.; Bronze, R.; Duarte, C.M.; Serra, A.T.; Pinto, R.; Freitas, M.; et al. Anti-inflammatory effect of rosmarinic acid and an extract of Rosmarinus officinalis in rat models of local and systemic inflammation. Basic Clin. Pharmacol. Toxicol. 2015, 116, 398–413. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht, J.; Hemmati, A.A.; Arzi, A.; Mansouri, M.T.; Rezaie, A.; Ghafourian, M. Protective effect of gallic acid against bleomycin-induced pulmonary fibrosis in rats. Pharmacol. Rep. 2015, 67, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Sittipunt, C.; Steinberg, K.P.; Ruzinski, J.T.; Myles, C.; Zhu, S.; Goodman, R.B.; Hudson, L.D.; Matalon, S.; Martin, T.R. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001, 163, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Hashimoto, S.; Kooguchi, K.; Kitamura, Y.; Onodera, H.; Urata, Y.; Ashihara, T. Expression of inducible nitric oxide synthase and inflammatory cytokines in alveolar macrophages of ARDS following sepsis. Chest 1998, 113, 1632–1639. [Google Scholar] [CrossRef]
- Redington, A.E.; Meng, Q.H.; Springall, D.R.; Evans, T.J.; Creminon, C.; Maclouf, J.; Holgate, S.T.; Howarth, P.H.; Polak, J.M. Increased expression of inducible nitric oxide synthase and cyclo-oxygenase-2 in the airway epithelium of asthmatic subjects and regulation by corticosteroid treatment. Thorax 2001, 56, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Maestrelli, P.; Paska, C.; Saetta, M.; Turato, G.; Nowicki, Y.; Monti, S.; Formichi, B.; Miniati, M.; Fabbri, L.M. Decreased haem oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients. Eur. Respir. J. 2003, 21, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Isakow, W.; Byers, D.E.; Engle, J.T.; Griffin, E.A.; Kemp, D.; Brody, S.L.; Gropler, R.J.; Miller, J.P.; Chu, W.; et al. Imaging pulmonary inducible nitric oxide synthase expression with PET. J. Nucl. Med. 2015, 56, 76–81. [Google Scholar] [CrossRef]
- Laudes, I.J.; Guo, R.F.; Riedemann, N.C.; Speyer, C.; Craig, R.; Sarma, J.V.; Ward, P.A. Disturbed homeostasis of lung intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 during sepsis. Am. J. Pathol. 2004, 164, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef]
- Budinger, G.R.; Mutlu, G.M.; Urich, D.; Soberanes, S.; Buccellato, L.J.; Hawkins, K.; Chiarella, S.E.; Radigan, K.A.; Eisenbart, J.; Agrawal, H.; et al. Epithelial cell death is an important contributor to oxidant-mediated acute lung injury. Am. J. Respir. Crit. Care Med. 2011, 183, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martinez-Caballero, S.; Osinska, H.; Cheng, E.H.; Robbins, J.; et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013, 2, e00772. [Google Scholar] [CrossRef] [PubMed]
- Husari, A.W.; Dbaibo, G.S.; Bitar, H.; Khayat, A.; Panjarian, S.; Nasser, M.; Bitar, F.F.; El-Sabban, M.; Zaatari, G.; Mroueh, S.M. Apoptosis and the activity of ceramide, Bax and Bcl-2 in the lungs of neonatal rats exposed to limited and prolonged hyperoxia. Respir. Res. 2006, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Lu, P.; Song, G.; Liu, Q.; Zhu, D.; Liu, X. Involvement of PI3K/Akt, ERK and p38 signaling pathways in emodin-mediated extrinsic and intrinsic human hepatoblastoma cell apoptosis. Food Chem. Toxicol. 2016, 92, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Martin, T.R. Science review: Apoptosis in acute lung injury. Crit. Care 2003, 7, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Valderrama, P.; Kaynar, A.M. The Basic Science and Molecular Mechanisms of Lung Injury and Acute Respiratory Distress Syndrome. Int. Anesthesiol. Clin. 2018, 56, 1–25. [Google Scholar] [CrossRef]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-kappaB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef]
- He, M.; Zhang, Y.; Xie, F.; Dou, X.; Han, M.; Zhang, H. Role of PI3K/Akt/NF-kappaB and GSK-3beta pathways in the rat model of cardiopulmonary bypass-related lung injury. Biomed. Pharmacother. 2018, 106, 747–754. [Google Scholar] [CrossRef]
- Vo, V.A.; Lee, J.W.; Kim, J.Y.; Park, J.H.; Lee, H.J.; Kim, S.S.; Kwon, Y.S.; Chun, W. Phosphorylation of Akt Mediates Anti-Inflammatory Activity of 1-p-Coumaroyl beta-D-Glucoside Against Lipopolysaccharide-Induced Inflammation in RAW264.7 Cells. Korean J. Physiol. Pharmacol. 2014, 18, 79–86. [Google Scholar] [CrossRef]
- Li, R.; Zou, X.; Huang, H.; Yu, Y.; Zhang, H.; Liu, P.; Pan, S.; Ouyang, Y.; Shang, Y. HMGB1/PI3K/Akt/mTOR Signaling Participates in the Pathological Process of Acute Lung Injury by Regulating the Maturation and Function of Dendritic Cells. Front. Immunol. 2020, 11, 1104. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Parkyn, L.; Otterbein, L.E.; Kureishi, Y.; Walsh, K.; Ray, A.; Ray, P. Activated Akt protects the lung from oxidant-induced injury and delays death of mice. J. Exp. Med. 2001, 193, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.F.; O’Connor, B. Proline specific peptidases. Biochim. Biophys. Acta 1997, 1343, 160–186. [Google Scholar] [CrossRef] [PubMed]
- Abdul Roda, M.; Sadik, M.; Gaggar, A.; Hardison, M.T.; Jablonsky, M.J.; Braber, S.; Blalock, J.E.; Redegeld, F.A.; Folkerts, G.; Jackson, P.L. Targeting prolyl endopeptidase with valproic acid as a potential modulator of neutrophilic inflammation. PLoS ONE 2014, 9, e97594. [Google Scholar] [CrossRef]
- Toppila, M.; Hytti, M.; Korhonen, E.; Ranta-Aho, S.; Harju, N.; Forsberg, M.M.; Kaarniranta, K.; Jalkanen, A.; Kauppinen, A. The Prolyl Oligopeptidase Inhibitor KYP-2047 Is Cytoprotective and Anti-Inflammatory in Human Retinal Pigment Epithelial Cells with Defective Proteasomal Clearance. Antioxidants 2023, 12, 1279. [Google Scholar] [CrossRef]
- Wells, J.M.; Jackson, P.L.; Viera, L.; Bhatt, S.P.; Gautney, J.; Handley, G.; King, R.W.; Xu, X.; Gaggar, A.; Bailey, W.C.; et al. A Randomized, Placebo-controlled Trial of Roflumilast. Effect on Proline-Glycine-Proline and Neutrophilic Inflammation in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 934–942. [Google Scholar] [CrossRef]
- Park, D.H.; Park, S.J.; Kim, J.M.; Jung, W.Y.; Ryu, J.H. Subchronic administration of rosmarinic acid, a natural prolyl oligopeptidase inhibitor, enhances cognitive performances. Fitoterapia 2010, 81, 644–648. [Google Scholar] [CrossRef]
- Huang, X.; Xiu, H.; Zhang, S.; Zhang, G. The Role of Macrophages in the Pathogenesis of ALI/ARDS. Mediat. Inflamm. 2018, 2018, 1264913. [Google Scholar] [CrossRef]
- Zundler, S.; Neurath, M.F. Interleukin-12: Functional activities and implications for disease. Cytokine Growth Factor Rev. 2015, 26, 559–568. [Google Scholar] [CrossRef]
- Maurya, H.; Mangal, V.; Gandhi, S.; Prabhu, K.; Ponnudurai, K. Prophylactic antioxidant potential of gallic Acid in murine model of sepsis. Int. J. Inflam. 2014, 2014, 580320. [Google Scholar] [CrossRef]
- Bastarache, J.A.; Blackwell, T.S. Development of animal models for the acute respiratory distress syndrome. Dis. Model Mech. 2009, 2, 218–223. [Google Scholar] [CrossRef]
- Kuperminc, E.; Heming, N.; Carlos, M.; Annane, D. Corticosteroids in ARDS. J. Clin. Med. 2023, 12, 3340. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Liao, X.; Wang, Y.; Chen, L.; Gao, W.; Wang, M.; Dai, H.; Yan, N.; Gao, Y.; Wu, X.; et al. Pharmacologic therapies of ARDS: From natural herb to nanomedicine. Front. Pharmacol. 2022, 13, 930593. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Singh, A.; Mishra, A. Gallic acid: Molecular rival of cancer. Environ. Toxicol. Pharmacol. 2013, 35, 473–485. [Google Scholar] [CrossRef]
- Gonzalez, R.F.; Dobbs, L.G. Isolation and culture of alveolar epithelial Type I and Type II cells from rat lungs. Methods Mol. Biol. 2013, 945, 145–159. [Google Scholar] [CrossRef]
- Liu, X.; Quan, N. Immune Cell Isolation from Mouse Femur Bone Marrow. Bio-Protocol 2015, 5, e1631. [Google Scholar] [CrossRef]
- Baldanzi, G.; Filigheddu, N.; Cutrupi, S.; Catapano, F.; Bonissoni, S.; Fubini, A.; Malan, D.; Baj, G.; Granata, R.; Broglio, F.; et al. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J. Cell Biol. 2002, 159, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Csonka, C.; Pali, T.; Bencsik, P.; Gorbe, A.; Ferdinandy, P.; Csont, T. Measurement of NO in biological samples. Br. J. Pharmacol. 2015, 172, 1620–1632. [Google Scholar] [CrossRef] [PubMed]
- Schuler, R.; Efentakis, P.; Wild, J.; Lagrange, J.; Garlapati, V.; Molitor, M.; Kossmann, S.; Oelze, M.; Stamm, P.; Li, H.; et al. T Cell-Derived IL-17A Induces Vascular Dysfunction via Perivascular Fibrosis Formation and Dysregulation of (.)NO/cGMP Signaling. Oxid. Med. Cell. Longev. 2019, 2019, 6721531. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Osteoarthr. Cartil. 2012, 20, 256–260. [Google Scholar] [CrossRef]
- Ding, H.; Ci, X.; Cheng, H.; Yu, Q.; Li, D. Chicoric acid alleviates lipopolysaccharide-induced acute lung injury in mice through anti-inflammatory and anti-oxidant activities. Int. Immunopharmacol. 2019, 66, 169–176. [Google Scholar] [CrossRef]
- Liang, Z.; Xu, Y.; Wen, X.; Nie, H.; Hu, T.; Yang, X.; Chu, X.; Yang, J.; Deng, X.; He, J. Rosmarinic Acid Attenuates Airway Inflammation and Hyperresponsiveness in a Murine Model of Asthma. Molecules 2016, 21, 769. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Gu, P.; Shen, H. Gallic acid improved inflammation via NF-kappaB pathway in TNBS-induced ulcerative colitis. Int. Immunopharmacol. 2019, 67, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, P.E.; Efentakis, P.; Abu Qourah, F.; Femmino, S.; Makridakis, M.; Kanaki, Z.; Varela, A.; Tsoumani, M.; Davos, C.H.; Dimitriou, C.A.; et al. Chronic Empagliflozin Treatment Reduces Myocardial Infarct Size in Nondiabetic Mice Through STAT-3-Mediated Protection on Microvascular Endothelial Cells and Reduction of Oxidative Stress. Antioxid. Redox Signal. 2021, 34, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Efentakis, P.; Kremastiotis, G.; Varela, A.; Nikolaou, P.E.; Papanagnou, E.D.; Davos, C.H.; Tsoumani, M.; Agrogiannis, G.; Konstantinidou, A.; Kastritis, E.; et al. Molecular mechanisms of carfilzomib-induced cardiotoxicity in mice and the emerging cardioprotective role of metformin. Blood 2019, 133, 710–723. [Google Scholar] [CrossRef]
- Tomashefski, J.F., Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin. Chest Med. 2000, 21, 435–466. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Garcia-Vicente, R.; Morales, M.L.; Ortiz-Ruiz, A.; Martinez-Lopez, J.; Linares, M. Protein Carbonylation and Lipid Peroxidation in Hematological Malignancies. Antioxidants 2020, 9, 1212. [Google Scholar] [CrossRef]
- Andreadou, I.; Iliodromitis, E.K.; Tsovolas, K.; Aggeli, I.K.; Zoga, A.; Gaitanaki, C.; Paraskevaidis, I.A.; Markantonis, S.L.; Beis, I.; Kremastinos, D.T. Acute administration of vitamin E triggers preconditioning via K(ATP) channels and cyclic-GMP without inhibiting lipid peroxidation. Free Radic. Biol. Med. 2006, 41, 1092–1099. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Product | Chemical Formula |
|---|---|
| Rosmarinic acid (RA) Ester of caffeic acid | 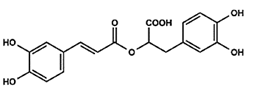 |
| Chicoric acid (CA) Hydroxycinnamic acid, phenylpropanoid | 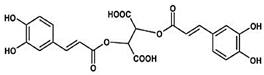 |
| Epigallocatechin-3-gallate (EGCG) Ester of epigallocatechin and gallic acid | 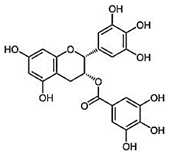 |
| Gallic acid (GA) Trihydroxybenzoic acid | 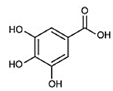 |
| Primer | Forward Sequence (5′–3′) | Reverse Sequence (5′–3′) | Product Length |
|---|---|---|---|
| β-actin | GCAAGCAGGAGTACGATGAGT | AGGGTGTAAAACGCAGCTCAG | 88 |
| Il-6 | AGTCCTTCCTACCCCAATTTCC | TGGTCTTGGTCCTTAGCCAC | 80 |
| Il-12α | CAAGGATATCTCTATGGTCAGCGT | GGTAGCGTGATTGACACATGC | 95 |
| Tnf-α | ATGGCCTCCCTCTCATCAGT | TGGTTTGCTACGACGTGGG | 100 |
| M-csf | CCTTCTTCGACATGGCTGGG | GTTCTGACACCTCCTTGGCA | 82 |
| Pop (Prep) | ACCTCCGTGCAGGAGTATCA | GTGCCTCCACGAAAGCCTTA | 97 |
| Ccl2 | CCAATGAGTAGGCTGGAGAGC | GAGCTTGGTGACAAAAACTACAGC | 81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zerikiotis, S.; Efentakis, P.; Dapola, D.; Agapaki, A.; Seiradakis, G.; Kostomitsopoulos, N.; Skaltsounis, A.-L.; Tseti, I.; Triposkiadis, F.; Andreadou, I. Synergistic Pulmonoprotective Effect of Natural Prolyl Oligopeptidase Inhibitors in In Vitro and In Vivo Models of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2023, 24, 14235. https://doi.org/10.3390/ijms241814235
Zerikiotis S, Efentakis P, Dapola D, Agapaki A, Seiradakis G, Kostomitsopoulos N, Skaltsounis A-L, Tseti I, Triposkiadis F, Andreadou I. Synergistic Pulmonoprotective Effect of Natural Prolyl Oligopeptidase Inhibitors in In Vitro and In Vivo Models of Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences. 2023; 24(18):14235. https://doi.org/10.3390/ijms241814235
Chicago/Turabian StyleZerikiotis, Stelios, Panagiotis Efentakis, Danai Dapola, Anna Agapaki, Georgios Seiradakis, Nikolaos Kostomitsopoulos, Alexios-Leandros Skaltsounis, Ioulia Tseti, Filippos Triposkiadis, and Ioanna Andreadou. 2023. "Synergistic Pulmonoprotective Effect of Natural Prolyl Oligopeptidase Inhibitors in In Vitro and In Vivo Models of Acute Respiratory Distress Syndrome" International Journal of Molecular Sciences 24, no. 18: 14235. https://doi.org/10.3390/ijms241814235