
Transcriptomic Analysis of the Response of Susceptible and Resistant Bitter Melon (Momordica charantia L.) to Powdery Mildew Infection Revealing Complex Resistance via Multiple Signaling Pathways

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
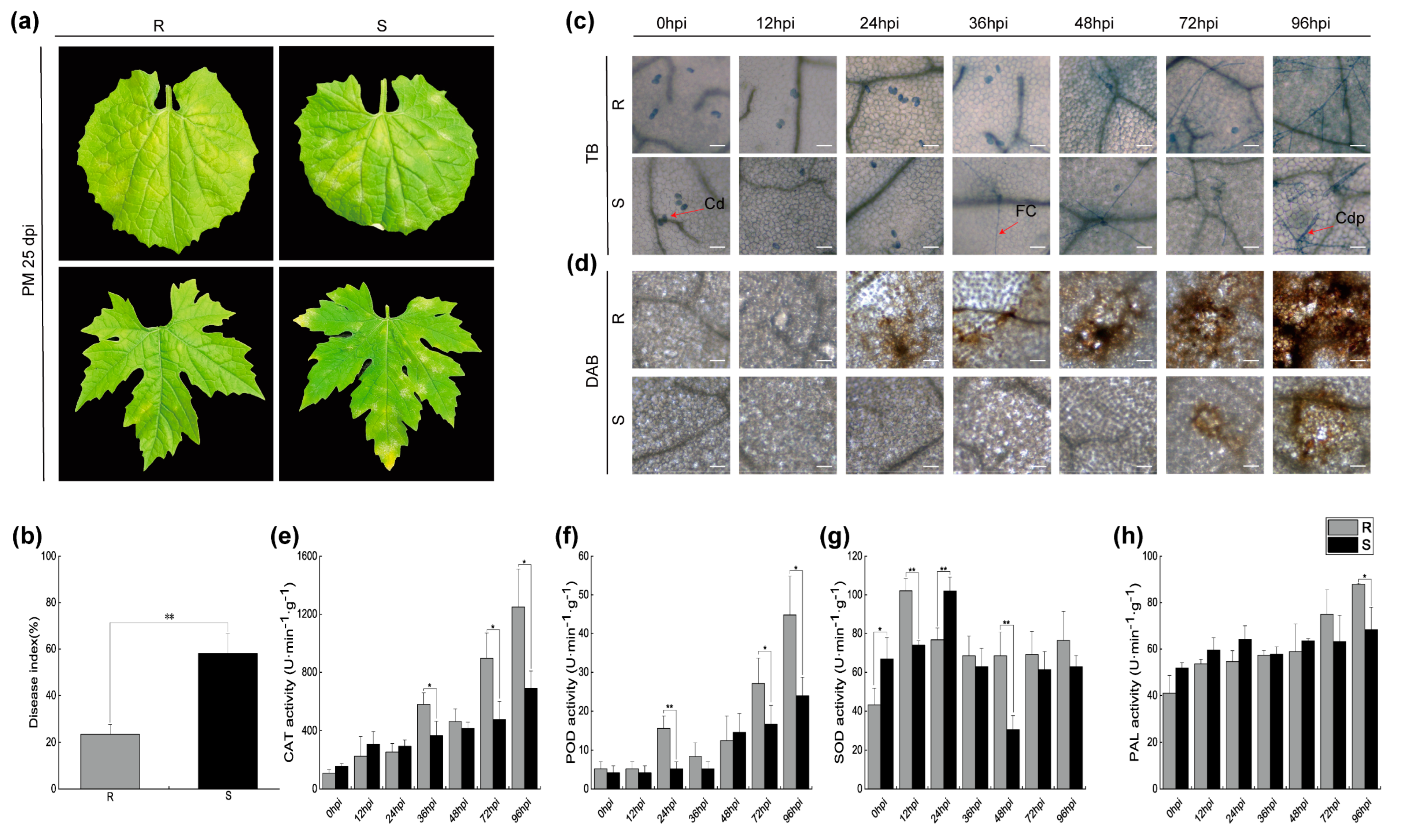
2.1. Symptoms and Physiological Changes of Bitter Melon Leaves Infected with P. xanthii
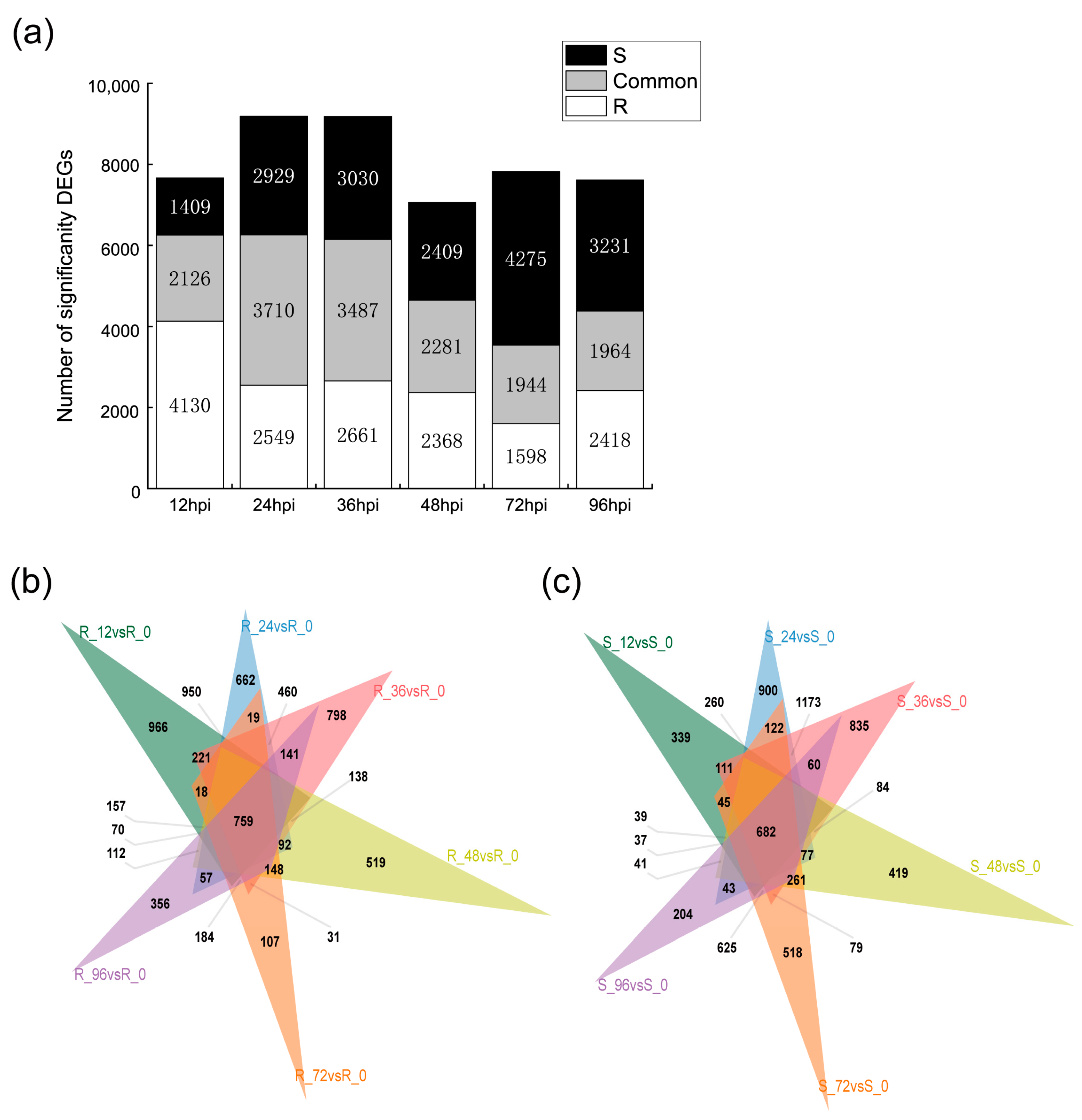
2.2. Transcriptomic Analysis of Resistant and Susceptible Bitter Melon Leaves in Response to P. xanthii at Different Time Points
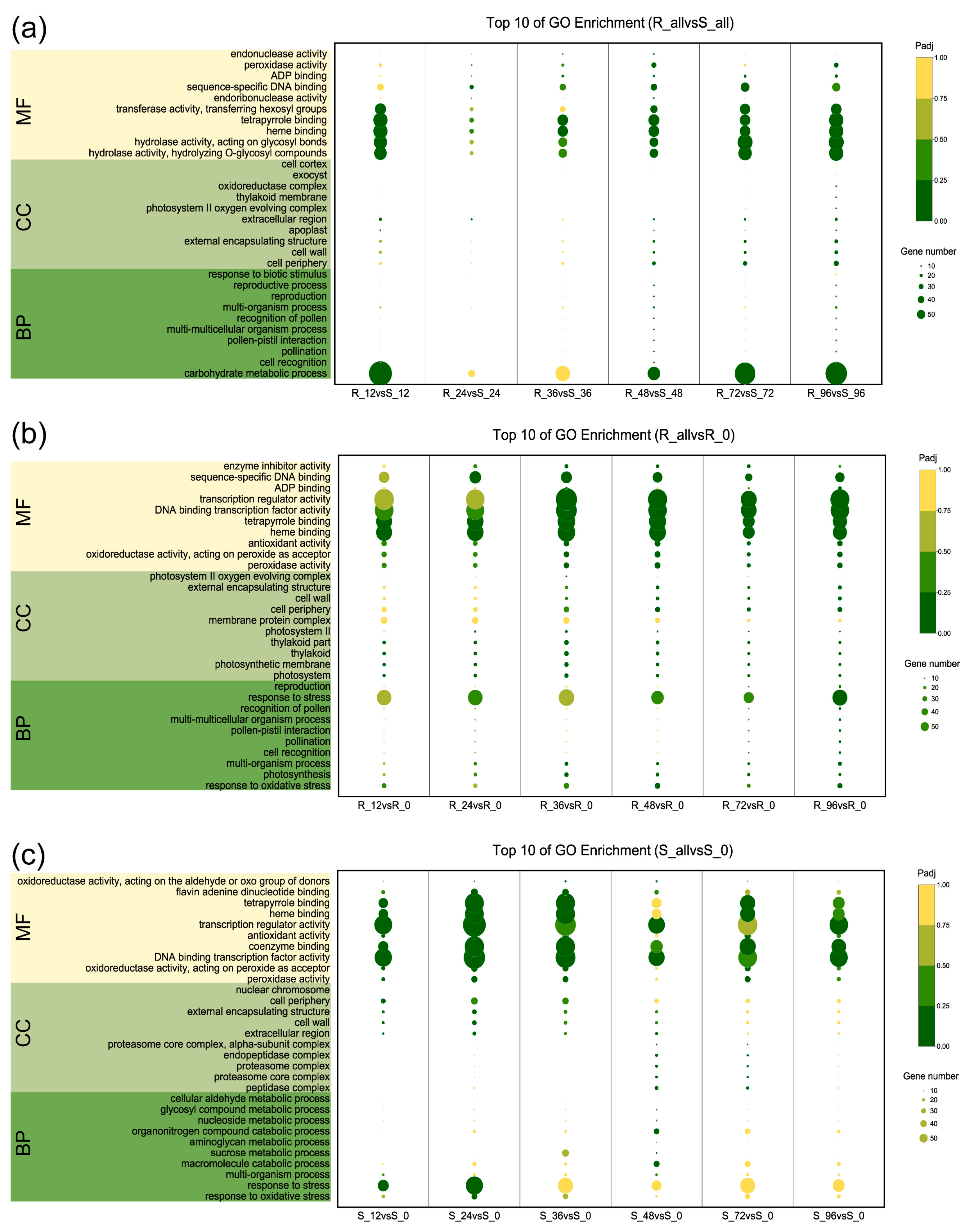
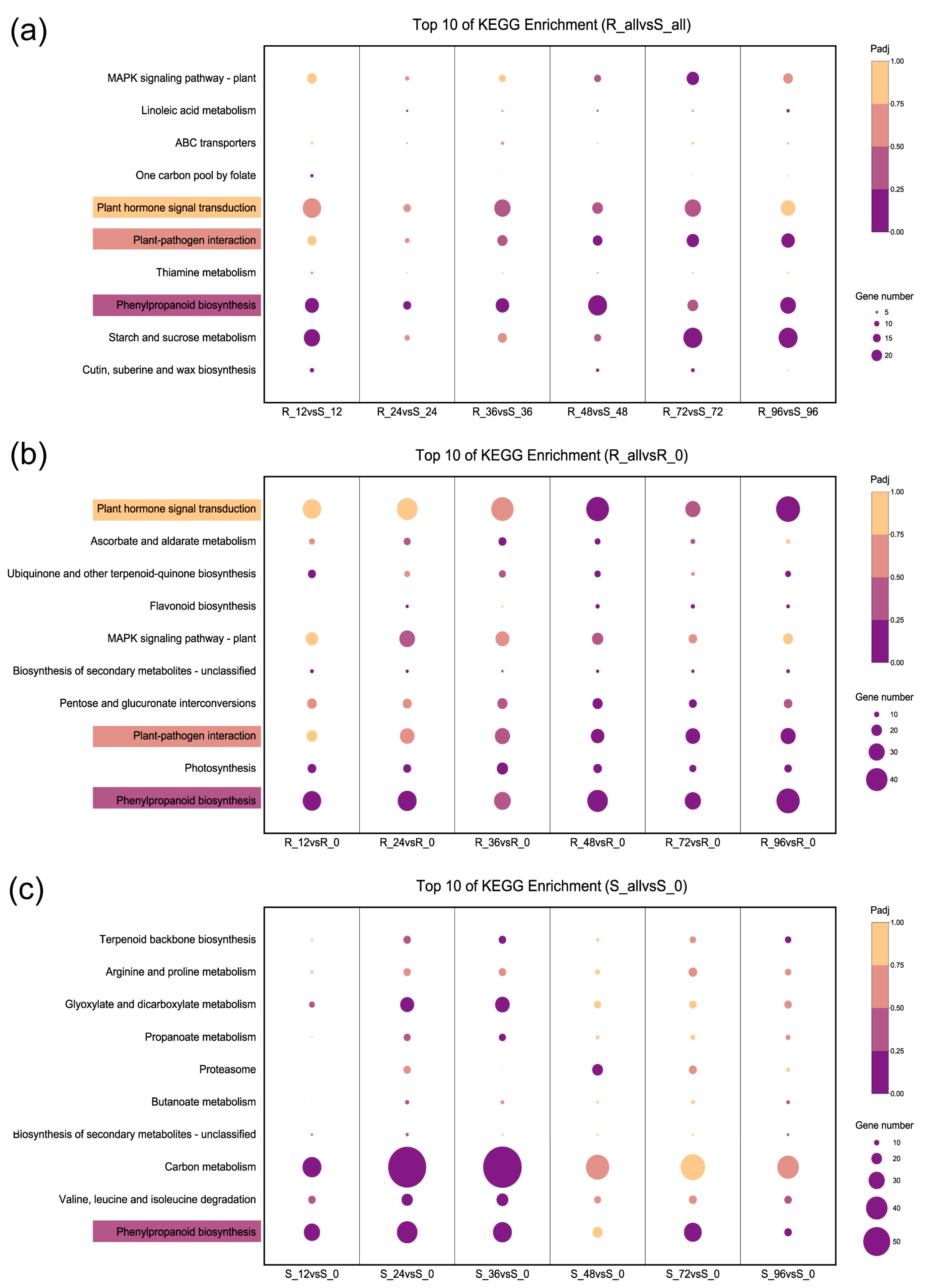
2.3. Functional Category Enrichment of Differentially Expressed Genes
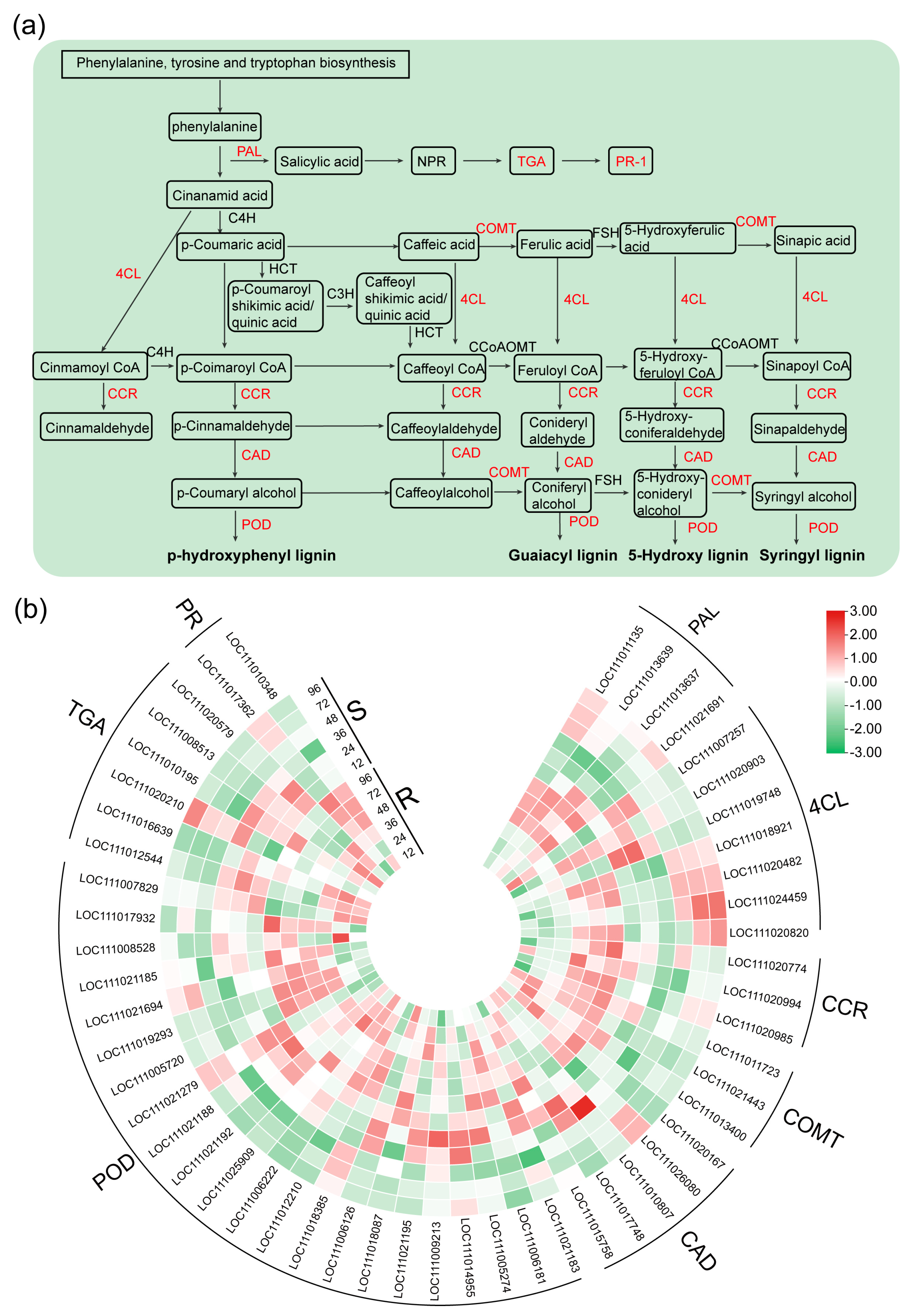
2.4. DEGs Related to the Phenylpropanoid Biosynthesis, SA Biosynthesis and Signaling Pathway
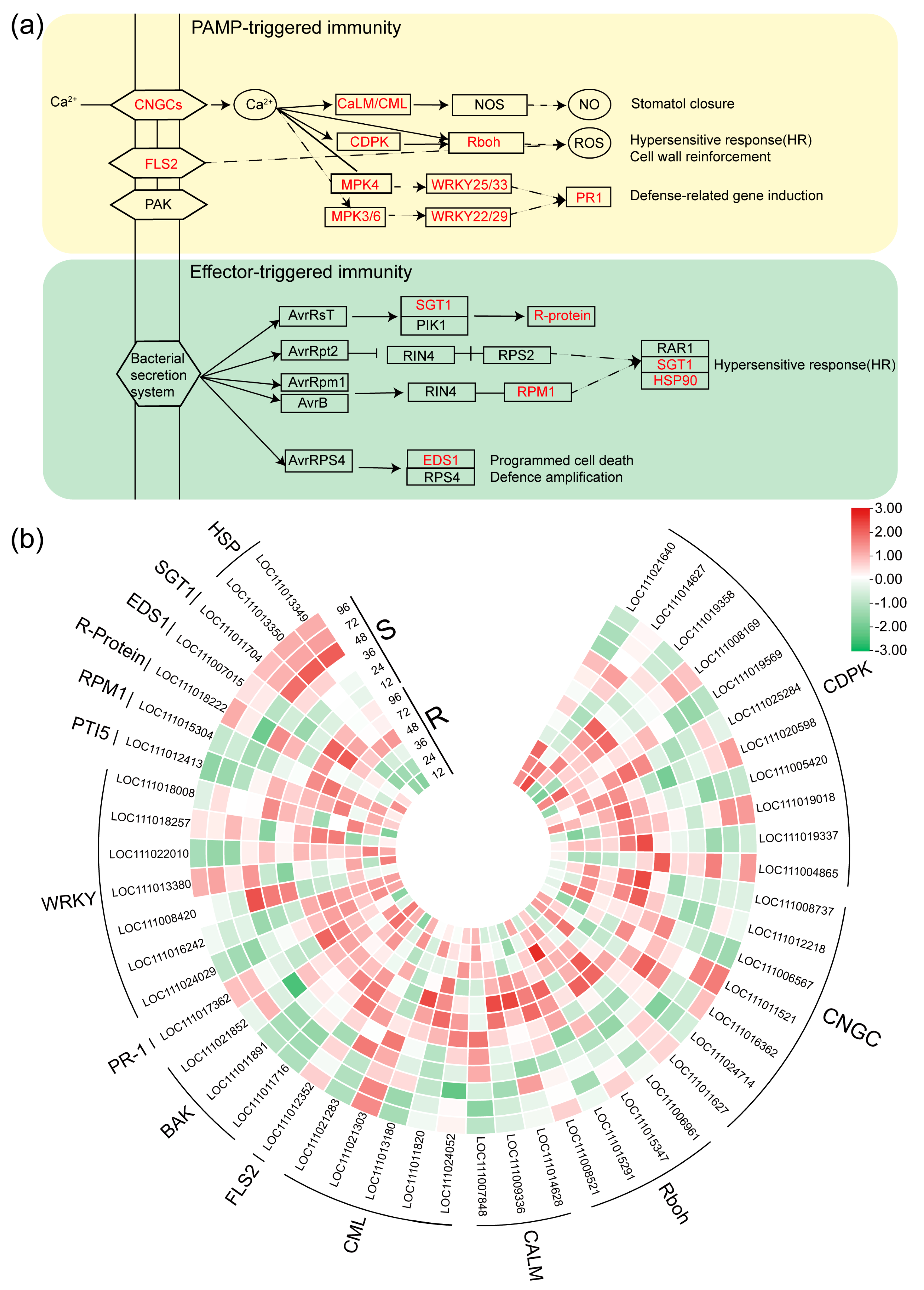
2.5. DEGs Related to the Plant-Pathogen Interaction Pathway
2.6. DEGs Related to the JA Biosynthesis and Signaling Pathway
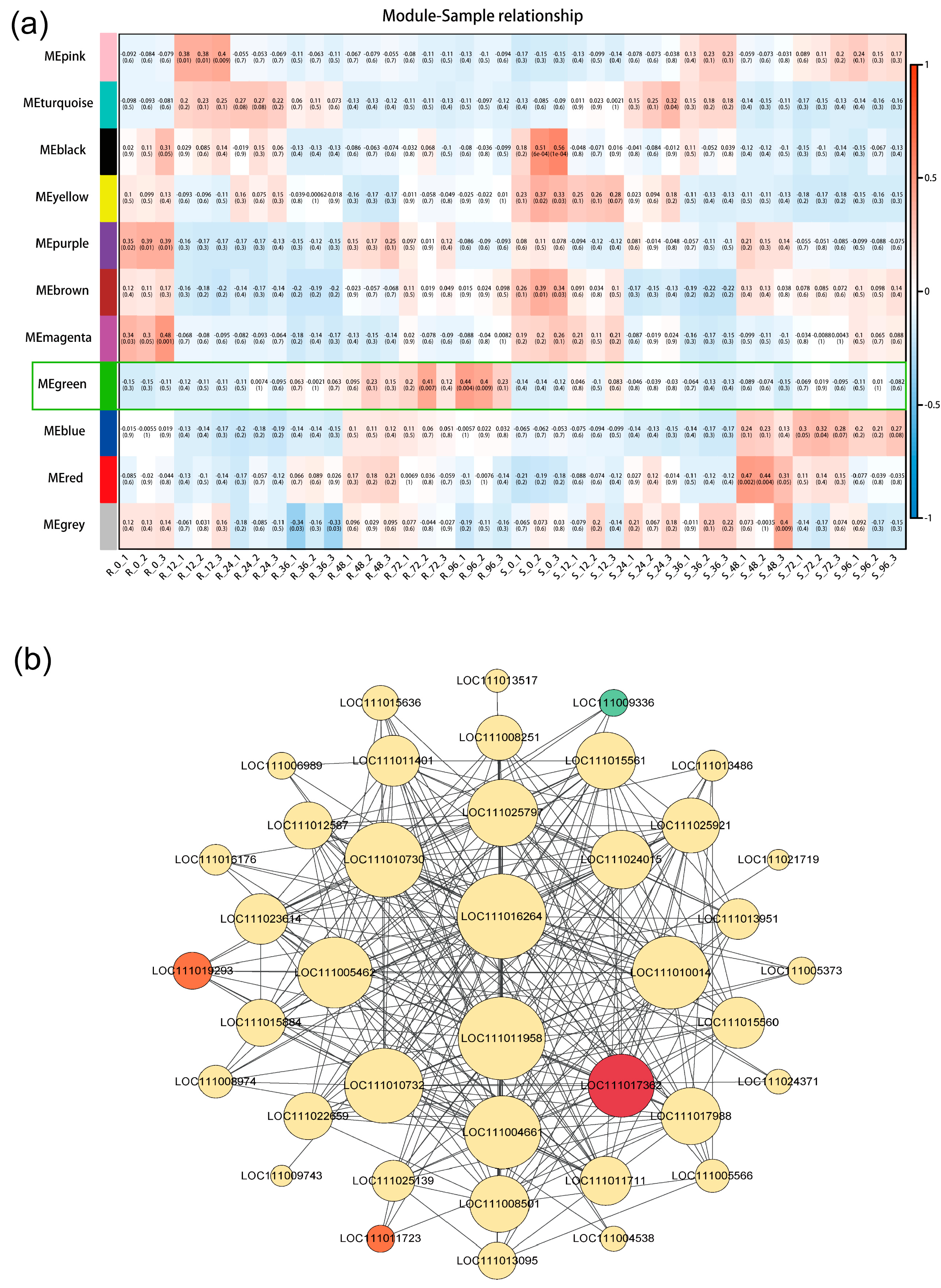
2.7. Identification of Key Hub Genes and Network Construction
2.8. Verification of DEGs Using Quantitative Reverse-Transcription PCR (qRT-PCR)
3. Discussion
4. Materials and Methods
4.1. Plant Materials and P. xanthii Inoculation
4.2. Investigation of Staining and Disease Index
4.3. Determination of Enzymes Activity
4.4. RNA Isolation and Sequencing
4.5. RNA-Seq Data Analysis
4.6. Construction of the Weighted Gene Co-Expression Network
4.7. qRT-PCR Validation
4.8. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dhillon, N.P.S.; Sanguansil, S.; Srimat, S.; Laenoi, S.; Schafleitner, R.; Pitrat, M.; McCreight, J.D. Inheritance of Resistance to Cucurbit Powdery Mildew in Bitter Gourd. HortScience 2019, 54, 1013–1016. [Google Scholar] [CrossRef]
- Behera, T.K.; Behera, S.; Bharathi, L.; John, K.J.; Simon, P.W.; Staub, J.E. Bitter gourd: Botany, horticulture, breeding. Hortic. Rev. 2010, 37, 101–141. [Google Scholar]
- Dandawate, P.R.; Subramaniam, D.; Padhye, S.B.; Anant, S. Bitter melon: A panacea for inflammation and cancer. Chin. J. Nat. Med. 2016, 14, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, W.; Gubler, W.; Grove, G. Epidemiology of powdery mildews in agricultural pathosystems. In The Powdery Mildews: A Comprehensive Treatise; Bélanger, R.R., Bushnell, W.R., Dik, A.J., Carver, T.L.W., Eds.; American Phytopathological Society (APS Press): St. Paul, MN, USA, 2002; pp. 169–199. [Google Scholar]
- Chen, Q.; Yu, G.; Wang, X.; Meng, X.; Lv, C. Genetics and resistance mechanism of the cucumber (Cucumis sativus L.) against powdery mildew. J. Plant Growth Regul. 2021, 40, 147–153. [Google Scholar] [CrossRef]
- Xin, M.; Wang, X.; Peng, H.; Yao, Y.; Xie, C.; Han, Y.; Ni, Z.; Sun, Q. Transcriptome comparison of susceptible and resistant wheat in response to powdery mildew infection. Genom. Proteom. Bioinform. 2012, 10, 94–106. [Google Scholar] [CrossRef]
- Cao, Y.; Diao, Q.; Lu, S.; Zhang, Y.; Yao, D. Comparative transcriptomic analysis of powdery mildew resistant and susceptible melon inbred lines to identify the genes involved in the response to Podosphaera xanthii infection. Sci. Hortic. 2022, 304, 111305. [Google Scholar] [CrossRef]
- Li, W.; Deng, Y.; Ning, Y.; He, Z.; Wang, G.-L. Exploiting broad-spectrum disease resistance in crops: From molecular dissection to breeding. Annu. Rev. Plant Biol. 2020, 71, 575–603. [Google Scholar] [CrossRef]
- Collum, T.D.; Culver, J.N. The impact of phytohormones on virus infection and disease. Curr. Opin. Virol. 2016, 17, 25–31. [Google Scholar] [CrossRef]
- Wang, D.; Dawadi, B.; Qu, J.; Ye, J. Light-engineering technology for enhancing plant disease resistance. Front. Plant Sci. 2022, 12, 805614. [Google Scholar] [CrossRef]
- Benjamin, G.; Pandharikar, G.; Frendo, P. Salicylic acid in plant symbioses: Beyond plant pathogen interactions. Biology 2022, 11, 861. [Google Scholar] [CrossRef]
- Fidler, J.; Graska, J.; Gietler, M.; Nykiel, M.; Prabucka, B.; Rybarczyk-Płońska, A.; Muszyńska, E.; Morkunas, I.; Labudda, M. PYR/PYL/RCAR receptors play a vital role in the abscisic-acid-dependent responses of plants to external or internal stimuli. Cells 2022, 11, 1352. [Google Scholar] [CrossRef] [PubMed]
- Ngou, B.P.M.; Jones, J.D.; Ding, P. Plant immune networks. Trends Plant Sci. 2022, 27, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gui, Y.; Li, Z.; Jiang, C.; Guo, J.; Niu, D. Induced systemic resistance for improving plant immunity by beneficial microbes. Plants 2022, 11, 386. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.; Kachroo, P. Fatty acid–derived signals in plant defense. Annu. Rev. Phytopathol. 2009, 47, 153–176. [Google Scholar] [CrossRef]
- Kouzai, Y.; Kimura, M.; Watanabe, M.; Kusunoki, K.; Osaka, D.; Suzuki, T.; Matsui, H.; Yamamoto, M.; Ichinose, Y.; Toyoda, K. Salicylic acid—Dependent immunity contributes to resistance against Rhizoctonia solani, a necrotrophic fungal agent of sheath blight, in rice and Brachypodium distachyon. New Phytol. 2018, 217, 771–783. [Google Scholar] [CrossRef]
- Shine, M.; Yang, J.W.; El-Habbak, M.; Nagyabhyru, P.; Fu, D.Q.; Navarre, D.; Ghabrial, S.; Kachroo, P.; Kachroo, A. Cooperative functioning between phenylalanine ammonia lyase and isochorismate synthase activities contributes to salicylic acid biosynthesis in soybean. New Phytol. 2016, 212, 627–636. [Google Scholar] [CrossRef]
- Bhuiyan, N.H.; Selvaraj, G.; Wei, Y.; King, J. Gene expression profiling and silencing reveal that monolignol biosynthesis plays a critical role in penetration defence in wheat against powdery mildew invasion. J. Exp. Bot. 2009, 60, 509–521. [Google Scholar] [CrossRef]
- Yadav, V.; Wang, Z.; Wei, C.; Amo, A.; Ahmed, B.; Yang, X.; Zhang, X. Phenylpropanoid pathway engineering: An emerging approach towards plant defense. Pathogens 2020, 9, 312. [Google Scholar] [CrossRef]
- Rong, W.; Luo, M.; Shan, T.; Wei, X.; Du, L.; Xu, H.; Zhang, Z. A wheat cinnamyl alcohol dehydrogenase TaCAD12 contributes to host resistance to the sharp eyespot disease. Front. Plant Sci. 2016, 7, 1723. [Google Scholar] [CrossRef]
- Yang, Q.; He, Y.; Kabahuma, M.; Chaya, T.; Kelly, A.; Borrego, E.; Bian, Y.; El Kasmi, F.; Yang, L.; Teixeira, P. A gene encoding maize caffeoyl-CoA O-methyltransferase confers quantitative resistance to multiple pathogens. Nat. Genet. 2017, 49, 1364–1372. [Google Scholar] [CrossRef]
- Zipfel, C.; Felix, G. Plants and animals: A different taste for microbes? Curr. Opin. Plant Biol. 2005, 8, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C. Pattern-recognition receptors in plant innate immunity. Curr. Opin. Immunol. 2008, 20, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.A.; Achnine, L.; Kota, P.; Liu, C.J.; Reddy, M.S.; Wang, L. The phenylpropanoid pathway and plant defence—A genomics perspective. Mol. Plant Pathol. 2002, 3, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Smigel, A.; Verma, R.; Berkowitz, G.A. Cyclic nucleotide gated channels and related signaling components in plant innate immunity. Plant Signal. Behav. 2009, 4, 277–282. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Shan, L.; Sheen, J. Elicitation and suppression of microbe-associated molecular pattern-triggered immunity in plant–microbe interactions. Cell. Microbiol. 2007, 9, 1385–1396. [Google Scholar] [CrossRef]
- Abramovitch, R.B.; Anderson, J.C.; Martin, G.B. Bacterial elicitation and evasion of plant innate immunity. Nat. Rev. Mol. Cell Biol. 2006, 7, 601–611. [Google Scholar] [CrossRef]
- Geng, S.; Li, A.; Tang, L.; Yin, L.; Wu, L.; Lei, C.; Guo, X.; Zhang, X.; Jiang, G.; Zhai, W. TaCPK2-A, a calcium-dependent protein kinase gene that is required for wheat powdery mildew resistance enhances bacterial blight resistance in transgenic rice. J. Exp. Bot. 2013, 64, 3125–3136. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, Y.; Yu, X.; Liu, J.; Yang, L.; Gao, Y.; Ke, G.; Zhou, M.; Mu, B.; Xiao, S. Overexpression of two CDPKs from wild Chinese grapevine enhances powdery mildew resistance in Vitis vinifera and Arabidopsis. New Phytol. 2021, 230, 2029–2046. [Google Scholar] [CrossRef]
- Hong, K.; Gong, D.; Zhang, L.; Hu, H.; Jia, Z.; Gu, H.; Song, K. Transcriptome characterization and expression profiles of the related defense genes in postharvest mango fruit against Colletotrichum gloeosporioides. Gene 2016, 576, 275–283. [Google Scholar] [CrossRef]
- Naveed, Z.A.; Ali, G.S. Comparative transcriptome analysis between a resistant and a susceptible wild tomato accession in response to Phytophthora parasitica. Int. J. Mol. Sci. 2018, 19, 3735. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Sun, M.; Jiang, Z.; Liu, Y.; Sun, Y.; Liu, D.; Jiang, C.; Ren, M.; Yuan, G.; Yu, W. Comparative transcriptome analysis reveals resistant and susceptible genes in tobacco cultivars in response to infection by Phytophthora nicotianae. Sci. Rep. 2021, 11, 809. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yu, Y.; Song, T.; Yu, Y.; Cui, N.; Ma, Z.; Chen, L.; Fan, H. Transcriptome sequence analysis of the defense responses of resistant and susceptible cucumber strains to Podosphaera xanthii. Front. Plant Sci. 2022, 13, 872218. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yan, W.; Yang, X.; Zhang, J.; Shi, Q. Comparative methylome reveals regulatory roles of DNA methylation in melon resistance to Podosphaera xanthii. Plant Sci. 2021, 309, 110954. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Wang, Z.; Guo, Y.; Zhang, X. Comparative transcriptome profiling reveals the role of phytohormones and phenylpropanoid pathway in early-stage resistance against powdery mildew in watermelon (Citrullus lanatus L.). Front. Plant Sci. 2022, 13, 1016822. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, L.; Yu, D. Wounding-induced WRKY8 is involved in basal defense in Arabidopsis. Mol. Plant-Microbe Interact. MPMI 2010, 23, 558–565. [Google Scholar] [CrossRef]
- Wasternack, C.; Song, S. Jasmonates: Biosynthesis, metabolism, and signaling by proteins activating and repressing transcription. J. Exp. Bot. 2017, 68, 1303–1321. [Google Scholar] [CrossRef]
- Bhuvnesh, K.; Pankaj, K.; Rajnish, S.; Arun, K. Regulatory interactions in phytohormone stress signaling implying plants resistance and resilience mechanisms. J. Plant Biochem. Biotechnol. 2021, 30, 813–828. [Google Scholar]
- Bozbuga, R. Expressions of Pathogenesis related 1 (PR1) Gene in Solanum lycopersicum and Influence of Salicylic Acid Exposures on Host-Meloidogyne incognita Interactions. Doklady Biochem. Biophys. 2020, 494, 266–269. [Google Scholar] [CrossRef]
- Sood, M.; Kapoor, D.; Kumar, V.; Kalia, N.; Bhardwaj, R.; Sidhu, G.P.S.; Sharma, A. Mechanisms of Plant Defense Under Pathogen Stress: A Review. Curr. Protein Pept. Sci. 2021, 22, 376–395. [Google Scholar] [CrossRef]
- Raji, M.R.; Lotfi, M.; Tohidfar, M.; Ramshini, H.; Sahebani, N.; Aalifar, M.; Baratian, M.; Mercati, F.; de Michele, R.; Carimi, F. Multiple fungal diseases resistance induction in Cucumis melo through co-transformation of different pathogenesis related (PR) protein genes. Sci. Hortic. 2022, 297, 110924. [Google Scholar] [CrossRef]
- Knoth, C.; Eulgem, T. The oomycete response gene LURP1 is required for defense against Hyaloperonospora parasitica in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2008, 55, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.Y.; Huang, Y.P. The signaling pathways of plant defense response and their interaction. Zhi Wu Sheng Li Yu Fen. Zi Sheng Wu Xue Xue Bao = J. Plant Physiol. Mol. Biol. 2005, 31, 347–353. [Google Scholar]
- Ha, C.M.; Rao, X.; Saxena, G.; Dixon, R.A. Growth-defense trade-offs and yield loss in plants with engineered cell walls. New Phytol. 2021, 231, 60–74. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, J.; Tschaplinski, T.J.; Tuskan, G.A.; Chen, J.G.; Muchero, W. Regulation of Lignin Biosynthesis and Its Role in Growth-Defense Tradeoffs. Front. Plant Sci. 2018, 9, 1427. [Google Scholar] [CrossRef]
- Cardoni, M.; Gómez-Lama Cabanás, C.; Valverde-Corredor, A.; Villar, R.; Mercado-Blanco, J. Unveiling Differences in Root Defense Mechanisms Between Tolerant and Susceptible Olive Cultivars to Verticillium dahliae. Front. Plant Sci. 2022, 13, 863055. [Google Scholar] [CrossRef]
- Gallego-Giraldo, L.; Posé, S.; Pattathil, S.; Peralta, A.G.; Hahn, M.G.; Ayre, B.G.; Sunuwar, J.; Hernandez, J.; Patel, M.; Shah, J.; et al. Elicitors and defense gene induction in plants with altered lignin compositions. New Phytol. 2018, 219, 1235–1251. [Google Scholar] [CrossRef]
- Sattler, S.E.; Funnell-Harris, D.L. Modifying lignin to improve bioenergy feedstocks: Strengthening the barrier against pathogens? Front. Plant Sci. 2013, 4, 70. [Google Scholar] [CrossRef]
- Bhuiyan, N.H.; Liu, W.; Liu, G.; Selvaraj, G.; Wei, Y.; King, J. Transcriptional regulation of genes involved in the pathways of biosynthesis and supply of methyl units in response to powdery mildew attack and abiotic stresses in wheat. Plant Mol. Biol. 2007, 64, 305–318. [Google Scholar] [CrossRef]
- Bhuiyan, N.H.; Selvaraj, G.; Wei, Y.; King, J. Role of lignification in plant defense. Plant Signal. Behav. 2009, 4, 158–159. [Google Scholar] [CrossRef]
- Zierold, U.; Scholz, U.; Schweizer, P. Transcriptome analysis of mlo-mediated resistance in the epidermis of barley. Mol. Plant Pathol. 2005, 6, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Junxin, Z.; Xihuan, Y.; Tiran, H.; Huan, L.; Fang, L.; Meixia, Y.; MingFeng, Y.; Lanqing, M. Overexpressing 4-coumaroyl-CoA ligase and stilbene synthase fusion genes in red raspberry plants leads to resveratrol accumulation and improved resistance against Botrytis cinerea. J. Plant Biochem. Biotechnol. 2022, 32, 85–91. [Google Scholar]
- Xiang, C.; Liu, J.; Ma, L.; Yang, M. Overexpressing codon-adapted fusion proteins of 4-coumaroyl-CoA ligase (4CL) and stilbene synthase (STS) for resveratrol production in Chlamydomonas reinhardtii. J. Appl. Phycol. 2020, 32, 1669–1676. [Google Scholar] [CrossRef]
- Chang, J.; Guo, Y.; Yan, J.; Zhang, Z.; Yuan, L.; Wei, C.; Zhang, Y.; Ma, J.; Yang, J.; Zhang, X.; et al. The role of watermelon caffeic acid O-methyltransferase (ClCOMT1) in melatonin biosynthesis and abiotic stress tolerance. Hortic. Res. 2021, 8, 210. [Google Scholar] [CrossRef]
- Tronchet, M.; Balagué, C.; Kroj, T.; Jouanin, L.; Roby, D. Cinnamyl alcohol dehydrogenases-C and D, key enzymes in lignin biosynthesis, play an essential role in disease resistance in Arabidopsis. Mol. Plant Pathol. 2010, 11, 83–92. [Google Scholar] [CrossRef]
- Kawasaki, T.; Koita, H.; Nakatsubo, T.; Hasegawa, K.; Wakabayashi, K.; Takahashi, H.; Umemura, K.; Umezawa, T.; Shimamoto, K. Cinnamoyl-CoA reductase, a key enzyme in lignin biosynthesis, is an effector of small GTPase Rac in defense signaling in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Almagro, L.; Gómez Ros, L.V.; Belchi-Navarro, S.; Bru, R.; Ros Barceló, A.; Pedreño, M.A. Class III peroxidases in plant defence reactions. J. Exp. Bot. 2009, 60, 377–390. [Google Scholar] [CrossRef]
- Garcia-Brugger, A.; Lamotte, O.; Vandelle, E.; Bourque, S.; Lecourieux, D.; Poinssot, B.; Wendehenne, D.; Pugin, A. Early signaling events induced by elicitors of plant defenses. Mol. Plant-Microbe Interact. MPMI 2006, 19, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Davis, L.C.; Verpoorte, R. Elicitor signal transduction leading to production of plant secondary metabolites. Biotechnol. Adv. 2005, 23, 283–333. [Google Scholar] [CrossRef]
- Cao, Y.; Diao, Q.; Chen, Y.; Jin, H.; Zhang, Y.; Zhang, H. Development of KASP Markers and Identification of a QTL Underlying Powdery Mildew Resistance in Melon (Cucumis melo L.) by Bulked Segregant Analysis and RNA-Seq. Front. Plant Sci. 2020, 11, 593207. [Google Scholar] [CrossRef]
- Liu, G.; Sheng, X.; Greenshields, D.L.; Ogieglo, A.; Kaminskyj, S.; Selvaraj, G.; Wei, Y. Profiling of wheat class III peroxidase genes derived from powdery mildew-attacked epidermis reveals distinct sequence-associated expression patterns. Mol. Plant-Microbe Interact. MPMI 2005, 18, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.; Deng, L.; Ruan, C.; Yi, L.; Zeng, K. Pichia galeiformis Induces Resistance in Postharvest Citrus by Activating the Phenylpropanoid Biosynthesis Pathway. J. Agric. Food Chem. 2021, 69, 2619–2631. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gao, P.; Wan, Y.; Cui, H.; Fan, C.; Liu, S.; Luan, F. Comparative transcriptome profiling of genes and pathways related to resistance against powdery mildew in two contrasting melon genotypes. Sci. Hortic. 2018, 227, 169–180. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, L.; Tu, L.; Liu, L.; Yuan, D.; Jin, L.; Long, L.; Zhang, X. Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J. Exp. Bot. 2011, 62, 5607–5621. [Google Scholar] [CrossRef] [PubMed]
- Lenardon, M.D.; Munro, C.A.; Gow, N.A. Chitin synthesis and fungal pathogenesis. Curr. Opin. Microbiol. 2010, 13, 416–423. [Google Scholar] [CrossRef]
- Moeder, W.; Urquhart, W.; Ung, H.; Yoshioka, K. The role of cyclic nucleotide-gated ion channels in plant immunity. Mol. Plant 2011, 4, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Ma, W.; Lemtiri-Chlieh, F.; Tsaltas, D.; Leng, Q.; von Bodman, S.; Berkowitz, G.A. Death don’t have no mercy and neither does calcium: Arabidopsis CYCLIC NUCLEOTIDE GATED CHANNEL2 and innate immunity. Plant Cell 2007, 19, 1081–1095. [Google Scholar] [CrossRef]
- Leba, L.J.; Cheval, C.; Ortiz-Martín, I.; Ranty, B.; Beuzón, C.R.; Galaud, J.P.; Aldon, D. CML9, an Arabidopsis calmodulin-like protein, contributes to plant innate immunity through a flagellin-dependent signalling pathway. Plant J. Cell Mol. Biol. 2012, 71, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Ali, G.S.; Celesnik, H.; Day, I.S. Coping with stresses: Roles of calcium- and calcium/calmodulin-regulated gene expression. Plant Cell 2011, 23, 2010–2032. [Google Scholar] [CrossRef]
- Boudsocq, M.; Sheen, J. CDPKs in immune and stress signaling. Trends Plant Sci. 2013, 18, 30–40. [Google Scholar] [CrossRef]
- Romeis, T.; Herde, M. From local to global: CDPKs in systemic defense signaling upon microbial and herbivore attack. Curr. Opin. Plant Biol. 2014, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gong, W.; Wang, Y.; Ma, Q. Contribution of a WRKY Transcription Factor, ShWRKY81, to Powdery Mildew Resistance in Wild Tomato. Int. J. Mol. Sci. 2023, 24, 2583. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, R.; Chao, J.; Lin, Y.E.; Jin, Q.; He, X.; Luo, S.; Wu, T. The expression patterns of Cucumis sativus WRKY (CsWRKY) family under the condition of inoculation with Phytophthora melonis in disease resistant and susceptible cucumber cultivars. Can. J. Plant Sci. 2015, 95, 1121–1131. [Google Scholar] [CrossRef]
- Xiao, S.; Brown, S.; Patrick, E.; Brearley, C.; Turner, J.G. Enhanced transcription of the Arabidopsis disease resistance genes RPW8. 1 and RPW8. 2 via a salicylic acid–dependent amplification circuit is required for hypersensitive cell death. Plant Cell 2003, 15, 33–45. [Google Scholar] [CrossRef]
- Sakata, Y.; Kubo, N.; Morishita, M.; Kitadani, E.; Sugiyama, M.; Hirai, M. QTL analysis of powdery mildew resistance in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2006, 112, 243–250. [Google Scholar] [CrossRef]
- Wang, N.; Wang, R.; Wang, R.; Chen, S. Transcriptomics analysis revealing candidate networks and genes for the body size sexual dimorphism of Chinese tongue sole (Cynoglossus semilaevis). Funct. Integr. Genom. 2018, 18, 327–339. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Zou, K.; Li, X.; Chen, F.; Cheng, Y.; Li, S.; Tian, L.; Shang, S. Transcriptomic Analysis of the Response of Susceptible and Resistant Bitter Melon (Momordica charantia L.) to Powdery Mildew Infection Revealing Complex Resistance via Multiple Signaling Pathways. Int. J. Mol. Sci. 2023, 24, 14262. https://doi.org/10.3390/ijms241814262
Chen X, Zou K, Li X, Chen F, Cheng Y, Li S, Tian L, Shang S. Transcriptomic Analysis of the Response of Susceptible and Resistant Bitter Melon (Momordica charantia L.) to Powdery Mildew Infection Revealing Complex Resistance via Multiple Signaling Pathways. International Journal of Molecular Sciences. 2023; 24(18):14262. https://doi.org/10.3390/ijms241814262
Chicago/Turabian StyleChen, Xuanyu, Kaixi Zou, Xuzhen Li, Feifan Chen, Yuyu Cheng, Shanming Li, Libo Tian, and Sang Shang. 2023. "Transcriptomic Analysis of the Response of Susceptible and Resistant Bitter Melon (Momordica charantia L.) to Powdery Mildew Infection Revealing Complex Resistance via Multiple Signaling Pathways" International Journal of Molecular Sciences 24, no. 18: 14262. https://doi.org/10.3390/ijms241814262
APA StyleChen, X., Zou, K., Li, X., Chen, F., Cheng, Y., Li, S., Tian, L., & Shang, S. (2023). Transcriptomic Analysis of the Response of Susceptible and Resistant Bitter Melon (Momordica charantia L.) to Powdery Mildew Infection Revealing Complex Resistance via Multiple Signaling Pathways. International Journal of Molecular Sciences, 24(18), 14262. https://doi.org/10.3390/ijms241814262