


Exploring the Interaction of New Pyridoquinazoline Derivatives with G-Quadruplex in the c-MYC Promoter Region

 ,
,  ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Antiproliferative Activity Evaluation
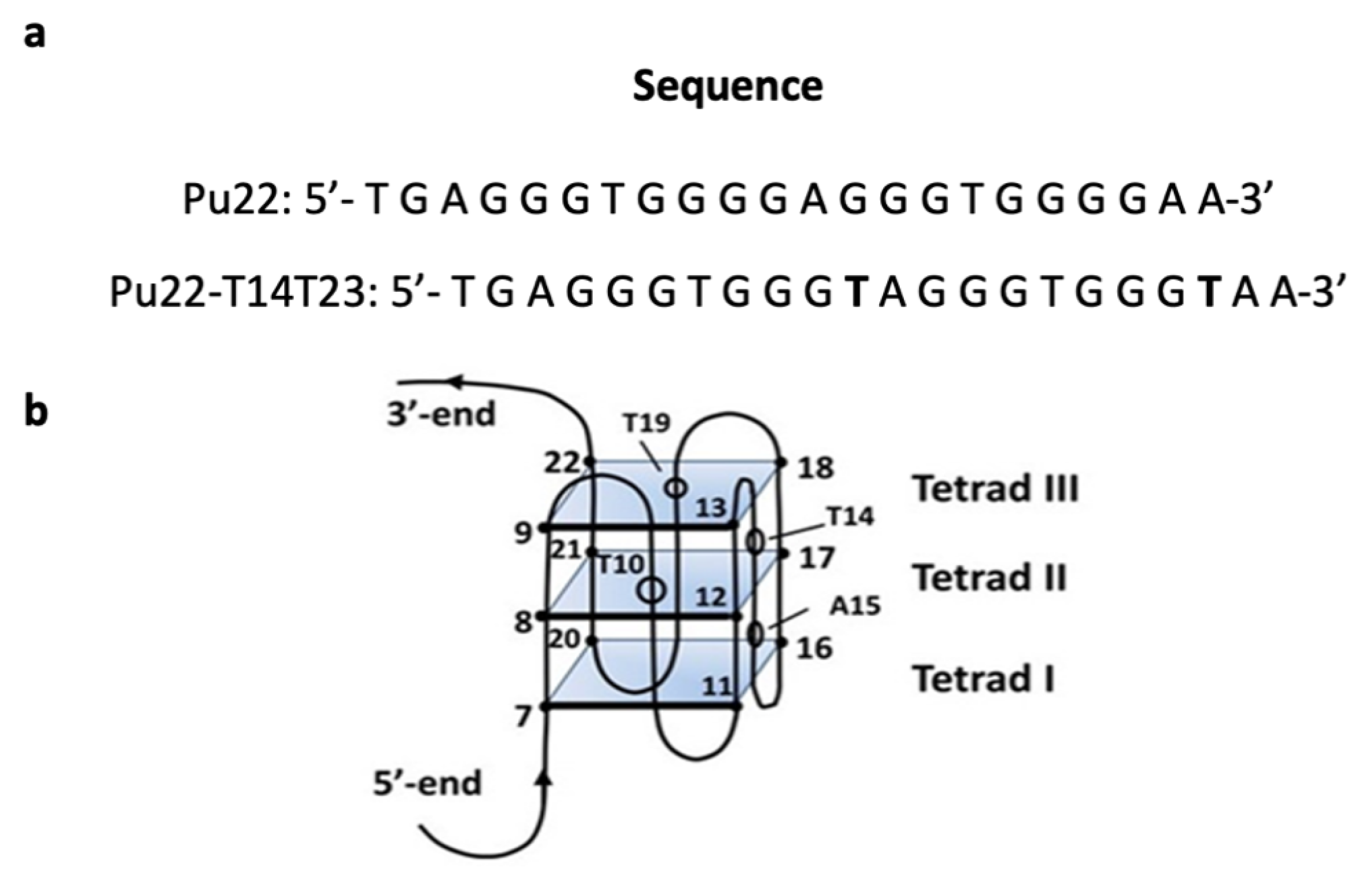
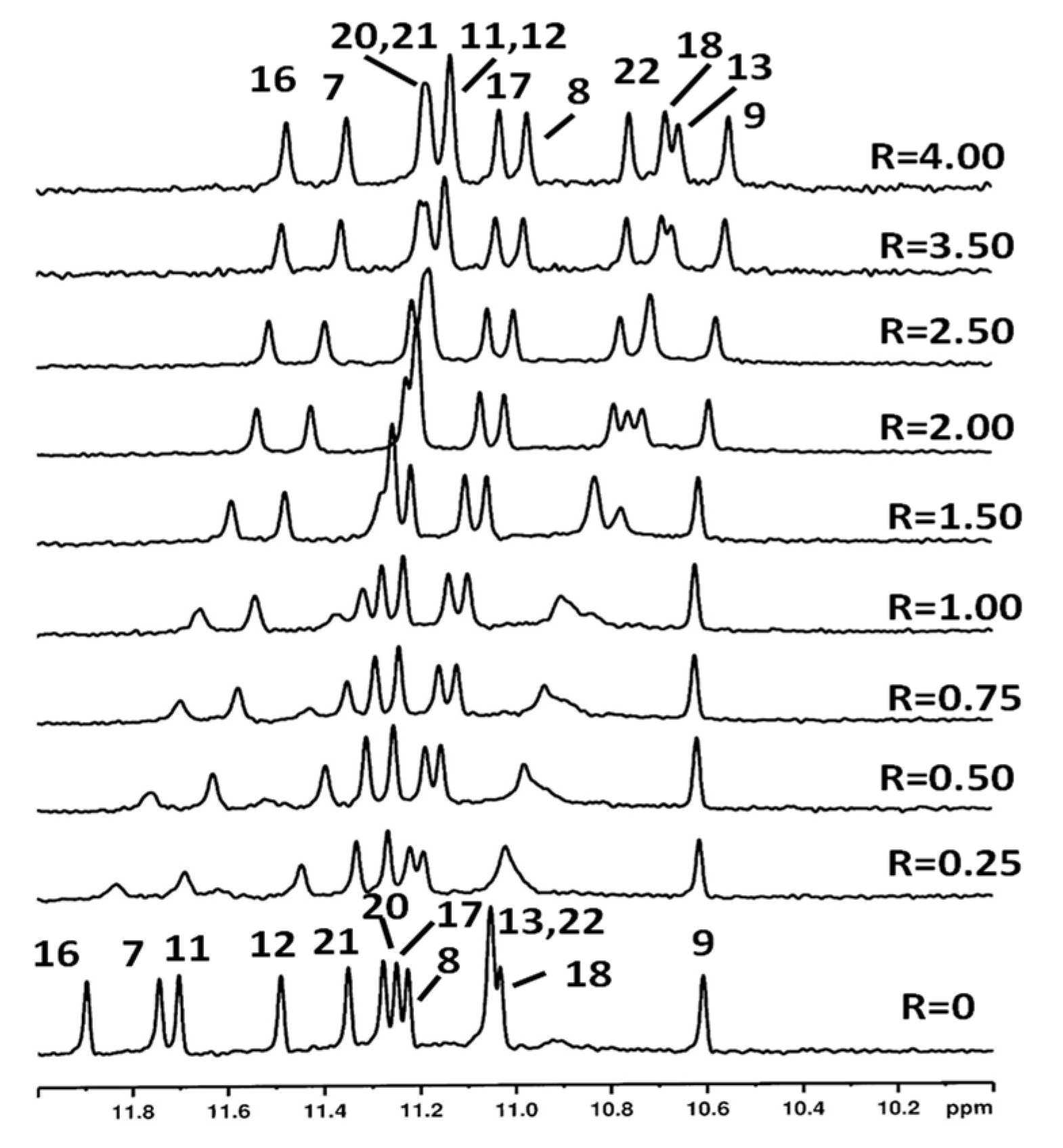
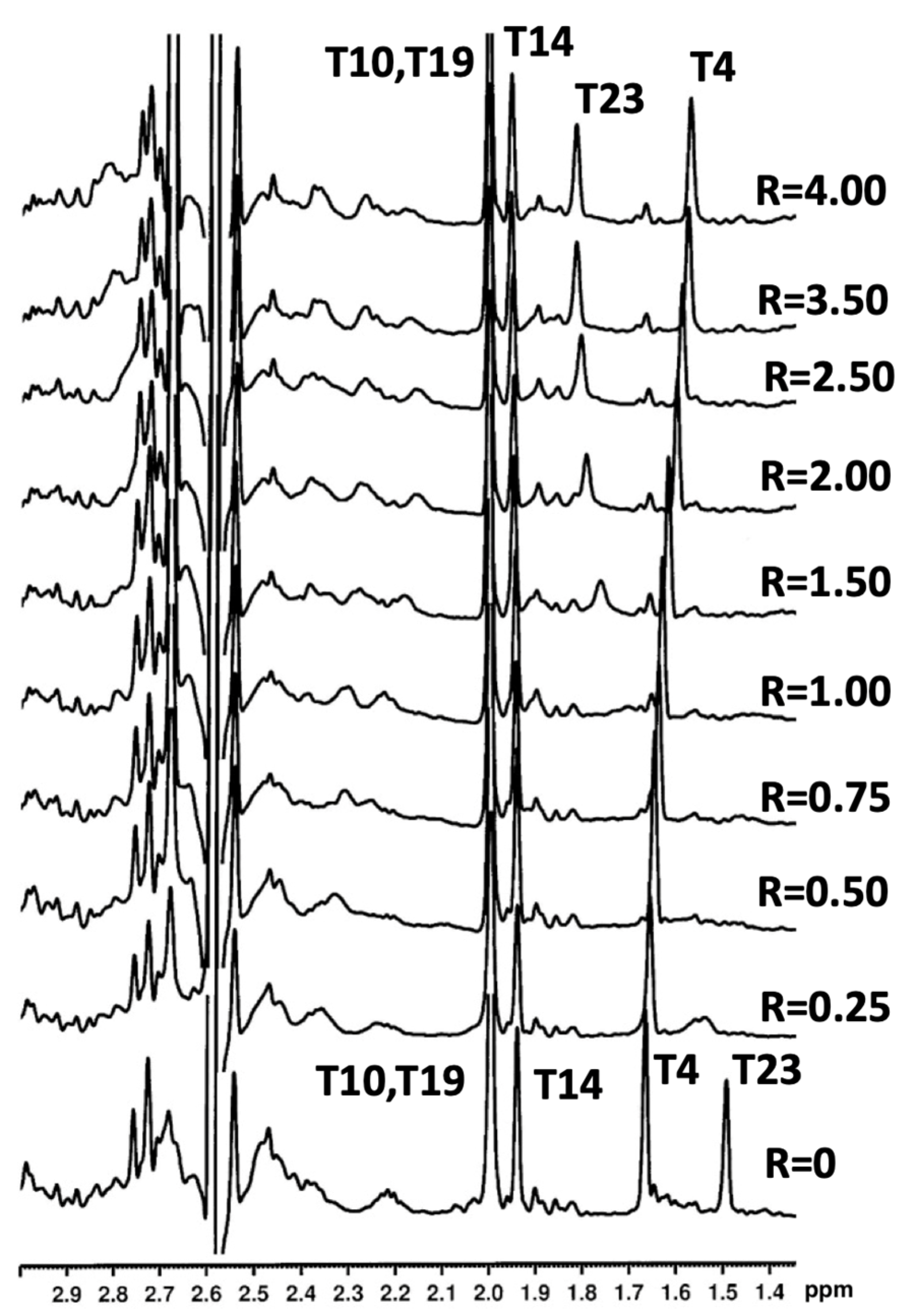
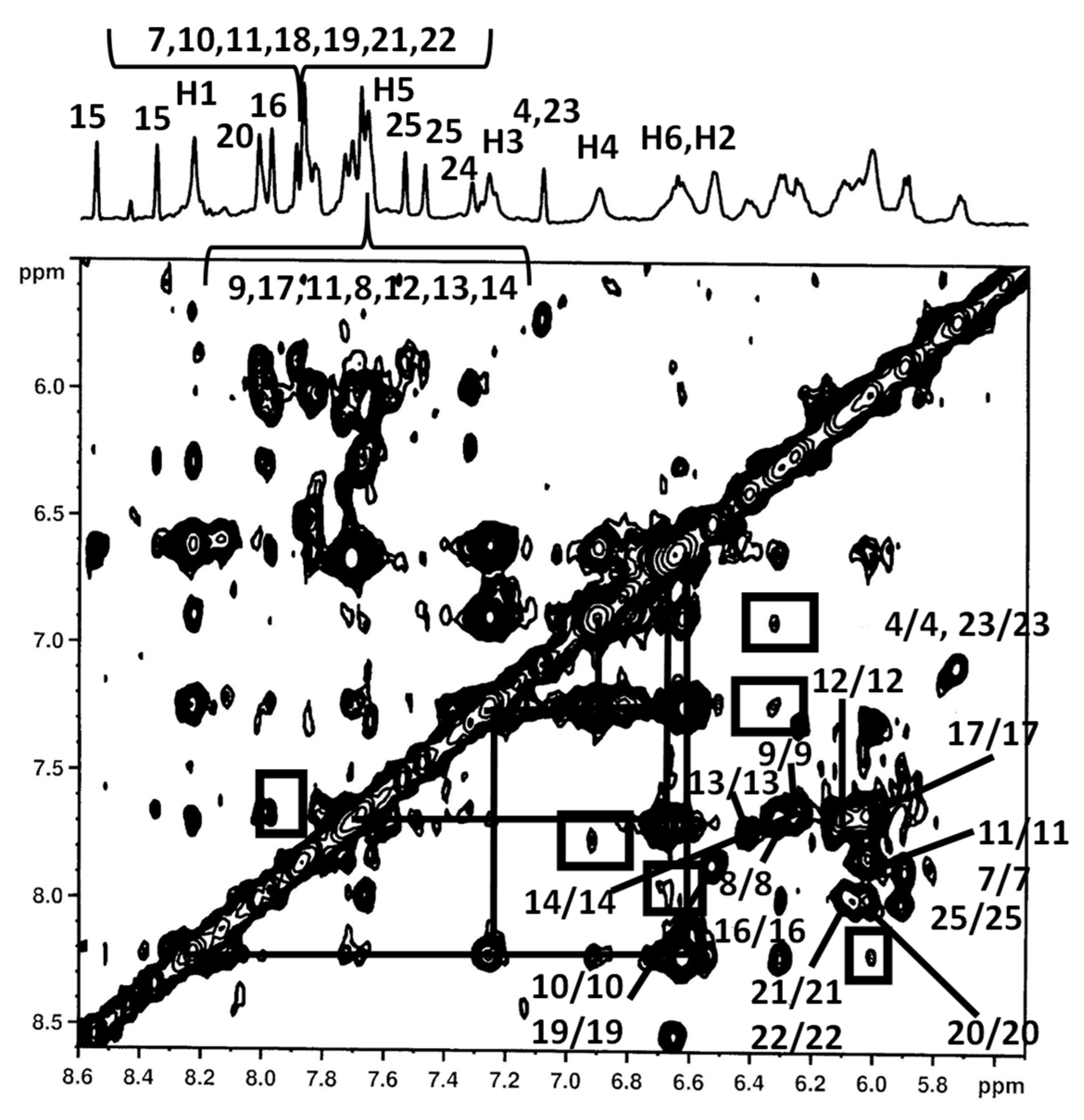
2.3. NMR Studies
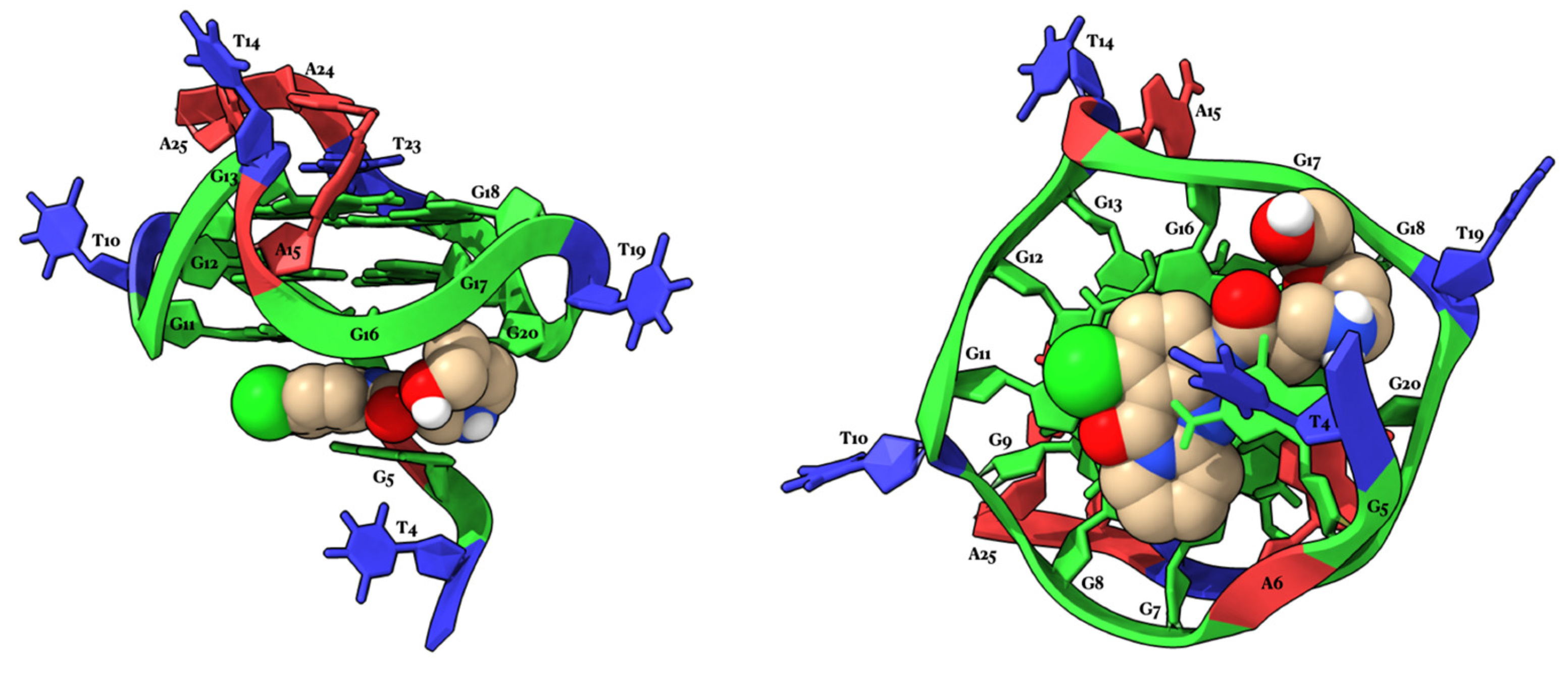
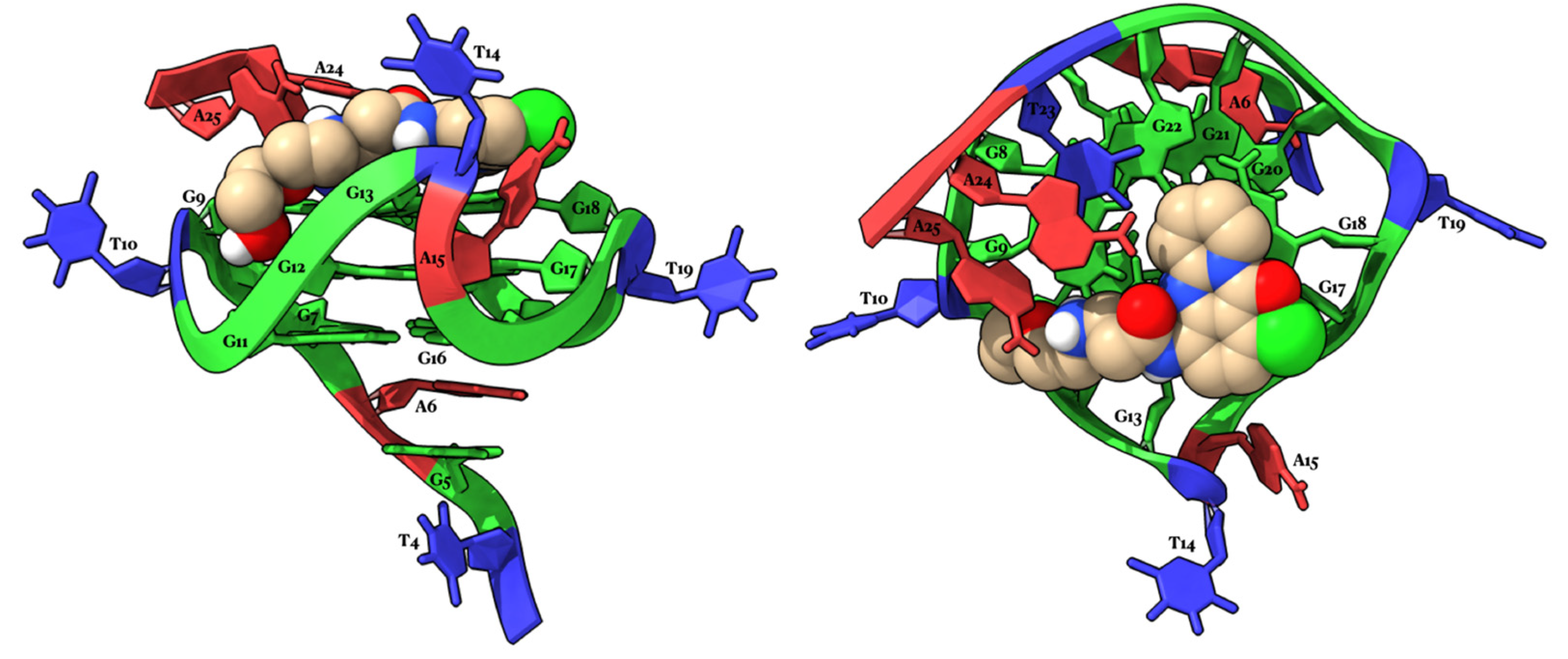
2.4. Molecular Modeling Studies
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Synthesis
3.2.1. Synthesis of 2,6-Dichloro-3-nitrobenzoic Acid (4)
3.2.2. Synthesis of 1-Chloro-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (6a) [21]
3.2.3. Synthesis of 1-Chloro-8-methoxy-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (6b)
3.2.4. Synthesis of 4-Amino-1-chloro-11H-pyrido[2,1-b]quinazolin-11-one (7a)
3.2.5. Synthesis of 4-Amino-1-chloro-8-methoxy-11H-pyrido[2,1-b]quinazolin-11-one (7b)
3.2.6. Synthesis of 2-Chloro-N-(1-chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (8a)
3.2.7. Synthesis of 2-Chloro-N-(1-chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (8b)
3.2.8. Synthesis of 3-Chloro-N-(1-chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (9a)
3.2.9. Synthesis of 3-Chloro-N-(1-chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (9b)
3.2.10. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-(dimethylamino)acetamide (1a)
3.2.11. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-(dimethylamino)acetamide (1b)
3.2.12. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-(diethylamino)acetamide (1c)
3.2.13. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-(diethylamino)acetamide (1d)
3.2.14. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-(4-methylpiperazin-1-yl)acetamide (1e)
3.2.15. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-(4-methylpiperazin-1-yl)acetamide (1f)
3.2.16. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-(4-(2-methoxyethyl)piperazin-1-yl)acetamide (1g)
3.2.17. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-(4-(2-methoxyethyl)piperazin-1-yl)acetamide (1h)
3.2.18. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-((2-(2-hydroxyethoxy)ethyl)amino)acetamide (1i)
3.2.19. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-2-((2-(2-hydroxyethoxy)ethyl)amino)acetamide (1j)
3.2.20. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-(dimethylamino)propanamide (2a)
3.2.21. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-(dimethylamino)propanamide (2b)
3.2.22. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-(diethylamino)propanamide (2c)
3.2.23. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-(diethylamino)propanamide (2d)
3.2.24. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-(4-methylpiperazin-1-yl)propanamide (2e)
3.2.25. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-(4-methylpiperazin-1-yl)propanamide (2f)
3.2.26. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-(4-(2-methoxyethyl)piperazin-1-yl)propanamide (2g)
3.2.27. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-(4-(2-methoxyethyl)piperazin-1-yl)propanamide (2h)
3.2.28. Synthesis of N-(1-Chloro-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-((2-(2-hydroxyethoxy)ethyl)amino)propanamide (2i)
3.2.29. Synthesis of N-(1-Chloro-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)-3-((2-(2-hydroxyethoxy)ethyl)amino)propanamide (2j)
3.3. NMR Experiments
3.4. Modeling Studies
3.5. Antiproliferative Activity Evaluation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cao, C.; Wang, X.; Yang, N.; Song, X.; Dong, X. Recent Advances of Cancer Chemodynamic Therapy Based on Fenton/Fenton-like Chemistry. Chem. Sci. 2022, 13, 863–889. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Chen, X.; Dobrovolskaia, M.A.; Lammers, T. Cancer Nanomedicine. Nat. Rev. Cancer 2022, 22, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Chaft, J.E.; Shyr, Y.; Sepesi, B.; Forde, P.M. Preoperative and Postoperative Systemic Therapy for Operable Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Montoya, S.; Soong, D.; Nguyen, N.; Affer, M.; Munamarty, S.P.; Taylor, J. Targeted Therapies in Cancer: To Be or Not to Be, Selective. Biomedicines 2021, 9, 1591. [Google Scholar] [CrossRef] [PubMed]
- Yahya, E.B.; Alqadhi, A.M. Recent Trends in Cancer Therapy: A Review on the Current State of Gene Delivery. Life Sci. 2021, 269, 119087. [Google Scholar] [CrossRef] [PubMed]
- Bahls, B.; Aljnadi, I.M.; Emídio, R.; Mendes, E.; Paulo, A. G-Quadruplexes in c-MYC Promoter as Targets for Cancer Therapy. Biomedicines 2023, 11, 969. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, H.; Qing, G. Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Modi, Y.S.; Patel, D.J. Propeller-Type Parallel-Stranded G-Quadruplexes in the Human c-Myc Promoter. J. Am. Chem. Soc. 2004, 126, 8710–8716. [Google Scholar] [CrossRef]
- Islam, M.A.; Thomas, S.D.; Murty, V.V.; Sedoris, K.J.; Miller, D.M. C-Myc Quadruplex-Forming Sequence Pu-27 Induces Extensive Damage in Both Telomeric and Nontelomeric Regions of DNA*. J. Biol. Chem. 2014, 289, 8521–8531. [Google Scholar] [CrossRef]
- Robinson, J.; Raguseo, F.; Nuccio, S.P.; Liano, D.; Di Antonio, M. DNA G-Quadruplex Structures: More than Simple Roadblocks to Transcription? Nucleic Acids Res. 2021, 49, 8419–8431. [Google Scholar] [CrossRef]
- Nakanishi, C.; Seimiya, H. G-Quadruplex in Cancer Biology and Drug Discovery. Biochem. Biophys. Res. Commun. 2020, 531, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-Quadruplexes: A Promising Target for Cancer Therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. G-Quadruplexes in Promoters throughout the Human Genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes. Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A Targeting Modality for Destruction of RNA Polymerase I That Possesses Anticancer Activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, K.; Colis, L.; Liu, H.; Jäämaa, S.; Moore, H.M.; Enbäck, J.; Laakkonen, P.; Vaahtokari, A.; Jones, R.J.; af Hällström, T.M.; et al. Identification of Novel P53 Pathway Activating Small-Molecule Compounds Reveals Unexpected Similarities with Known Therapeutic Agents. PLoS ONE 2010, 5, e12996. [Google Scholar] [CrossRef]
- Musso, L.; Mazzini, S.; Rossini, A.; Castagnoli, L.; Scaglioni, L.; Artali, R.; Di Nicola, M.; Zunino, F.; Dallavalle, S. C-MYC G-Quadruplex Binding by the RNA Polymerase I Inhibitor BMH-21 and Analogues Revealed by a Combined NMR and Biochemical Approach. Biochim. Biophys. Acta (BBA)-General. Subj. 2018, 1862, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Colis, L.; Ernst, G.; Sanders, S.; Liu, H.; Sirajuddin, P.; Peltonen, K.; DePasquale, M.; Barrow, J.C.; Laiho, M. Design, Synthesis, and Structure–Activity Relationships of Pyridoquinazolinecarboxamides as RNA Polymerase I Inhibitors. J. Med. Chem. 2014, 57, 4950–4961. [Google Scholar] [CrossRef]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution Structure of the Biologically Relevant G-Quadruplex Element in the Human c-MYC Promoter. Implications for G-Quadruplex Stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef]
- Dai, J.; Carver, M.; Hurley, L.H.; Yang, D. Solution Structure of a 2:1 Quindoline–c-MYC G-Quadruplex: Insights into G-Quadruplex-Interactive Small Molecule Drug Design. J. Am. Chem. Soc. 2011, 133, 17673–17680. [Google Scholar] [CrossRef]
- Pacheco-Benichou, A.; Ivendengani, E.; Kostakis, I.K.; Besson, T.; Fruit, C. Copper-Catalyzed C–H Arylation of Fused-Pyrimidinone Derivatives Using Diaryliodonium Salts. Catalysts 2021, 11, 28. [Google Scholar] [CrossRef]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.-P.; Ren, P.; Ponder, J.W. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative Partial Equalization of Orbital Electronegativity—A Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Harrach, M.F.; Drossel, B. Structure and Dynamics of TIP3P, TIP4P, and TIP5P Water near Smooth and Atomistic Walls of Different Hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A Refined Force Field for DNA Simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef]
- Lamoureux, G.; Roux, B. Modeling Induced Polarization with Classical Drude Oscillators: Theory and Molecular Dynamics Simulation Algorithm. J. Chem. Phys. 2003, 119, 3025–3039. [Google Scholar] [CrossRef]
- Jiang, W.; Hardy, D.J.; Phillips, J.C.; MacKerell, A.D.; Schulten, K.; Roux, B. High-Performance Scalable Molecular Dynamics Simulations of a Polarizable Force Field Based on Classical Drude Oscillators in NAMD. J. Phys. Chem. Lett. 2011, 2, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 44130. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Amato, J.; Miglietta, G.; Morigi, R.; Iaccarino, N.; Locatelli, A.; Leoni, A.; Novellino, E.; Pagano, B.; Capranico, G.; Randazzo, A. Monohydrazone Based G-Quadruplex Selective Ligands Induce DNA Damage and Genome Instability in Human Cancer Cells. J. Med. Chem. 2020, 63, 3090–3103. [Google Scholar] [CrossRef]
- Hu, M.-H.; Wang, Y.-Q.; Yu, Z.-Y.; Hu, L.-N.; Ou, T.-M.; Chen, S.-B.; Huang, Z.-S.; Tan, J.-H. Discovery of a New Four-Leaf Clover-Like Ligand as a Potent c-MYC Transcription Inhibitor Specifically Targeting the Promoter G-Quadruplex. J. Med. Chem. 2018, 61, 2447–2459. [Google Scholar] [CrossRef]
- Zizza, P.; Cingolani, C.; Artuso, S.; Salvati, E.; Rizzo, A.; D’Angelo, C.; Porru, M.; Pagano, B.; Amato, J.; Randazzo, A.; et al. Intragenic G-Quadruplex Structure Formed in the Human CD133 and Its Biological and Translational Relevance. Nucleic Acids Res. 2016, 44, 1579–1590. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R1 | R2 | IC50 (μΜ) (±) SD a |
|---|---|---|---|
 | - | - | 11.67 ± 1.53 |
| BMH21 | - | - | 0.45 ± 0.02 |
| 1a | -H | 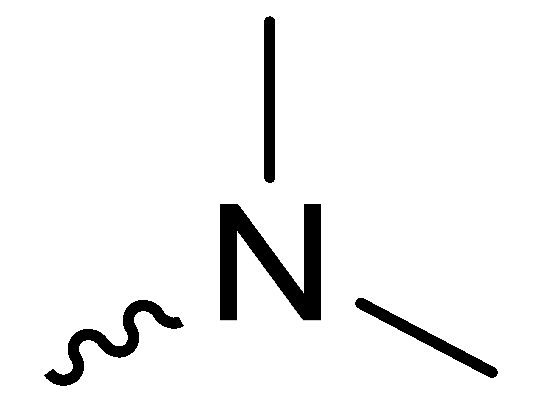 | >100 |
| 1b | -OCH3 |  | >100 |
| 1c | -H | 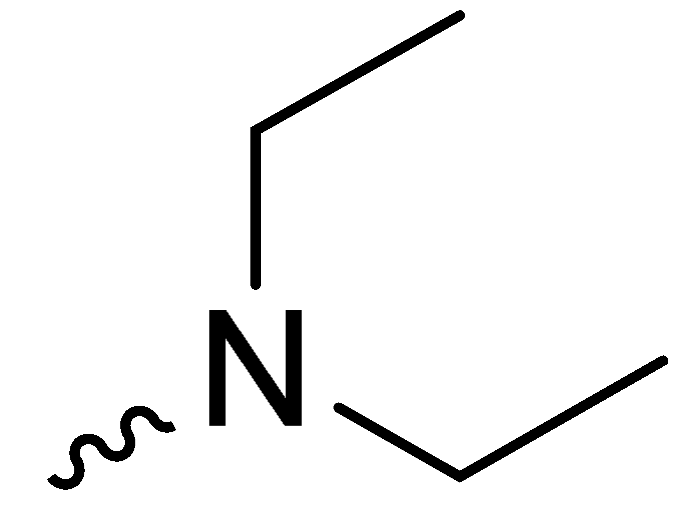 | 34.00 ± 1.00 |
| 1d | -OCH3 | 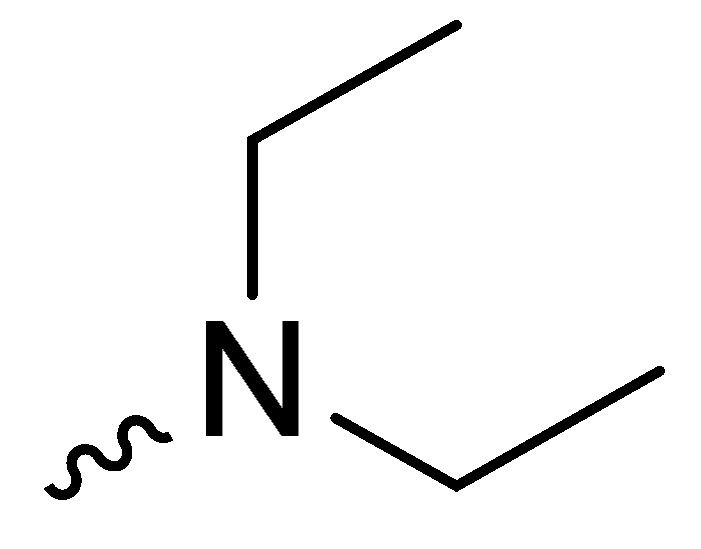 | NT * |
| 1e | -H | 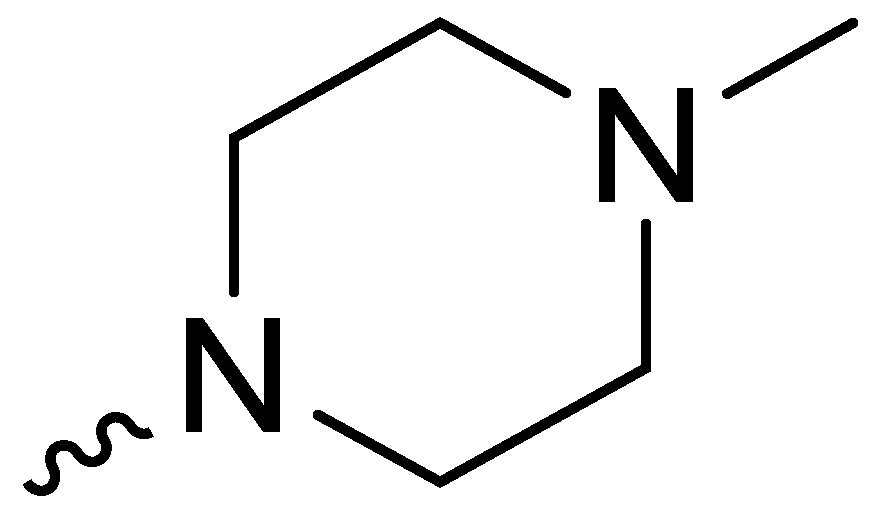 | NT * |
| 1f | -OCH3 |  | NT * |
| 1g | -H | 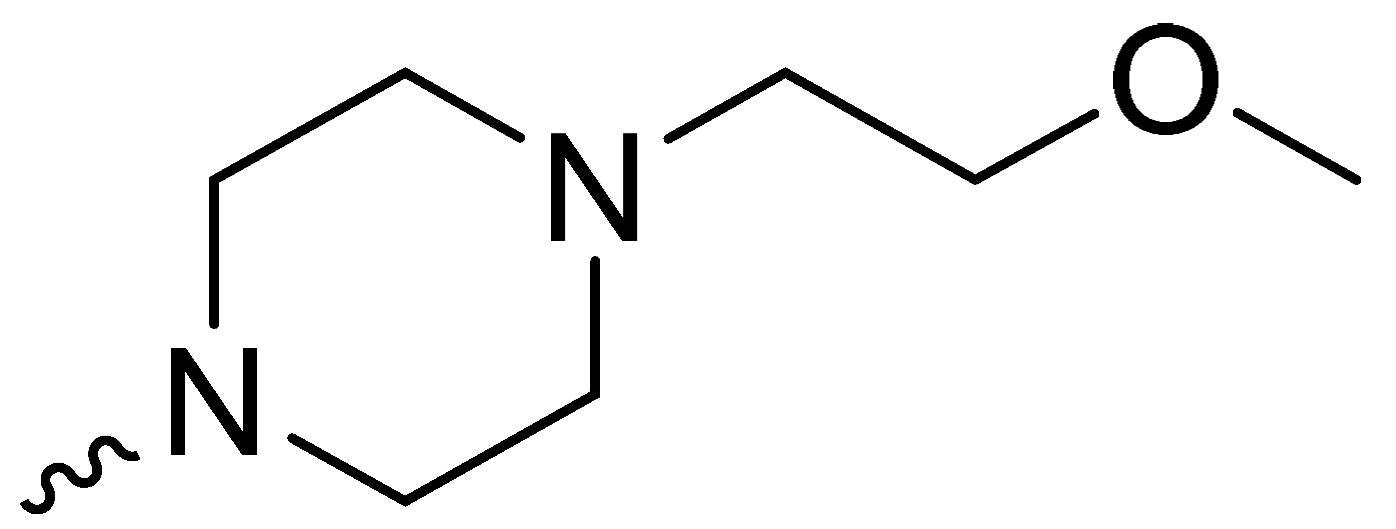 | NT * |
| 1h | -OCH3 | 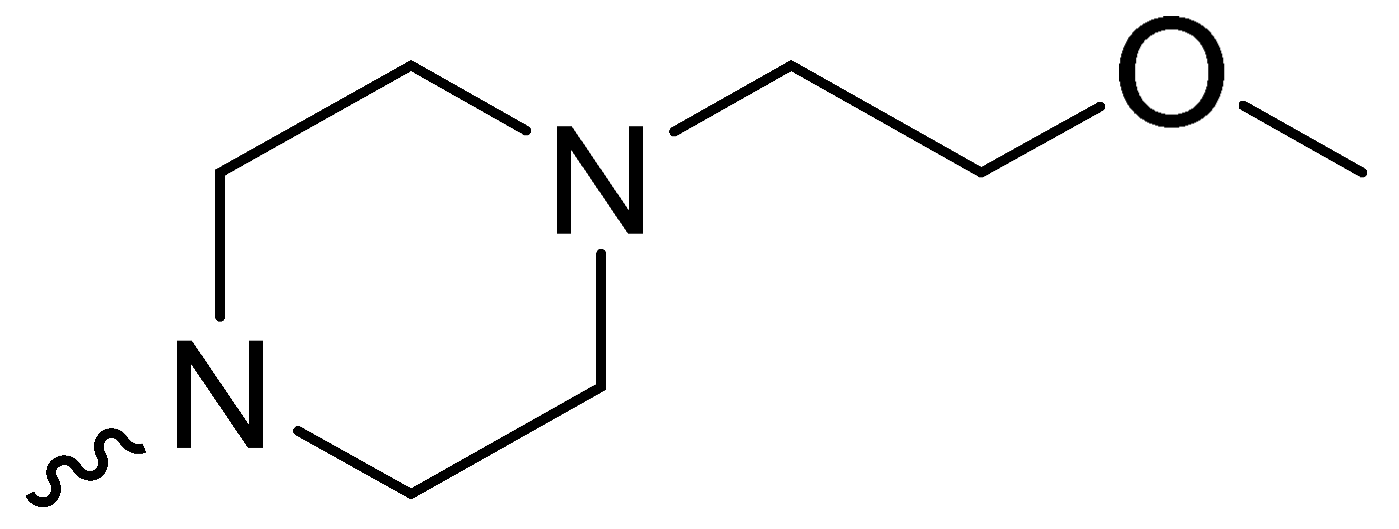 | NT * |
| 1i | -H | 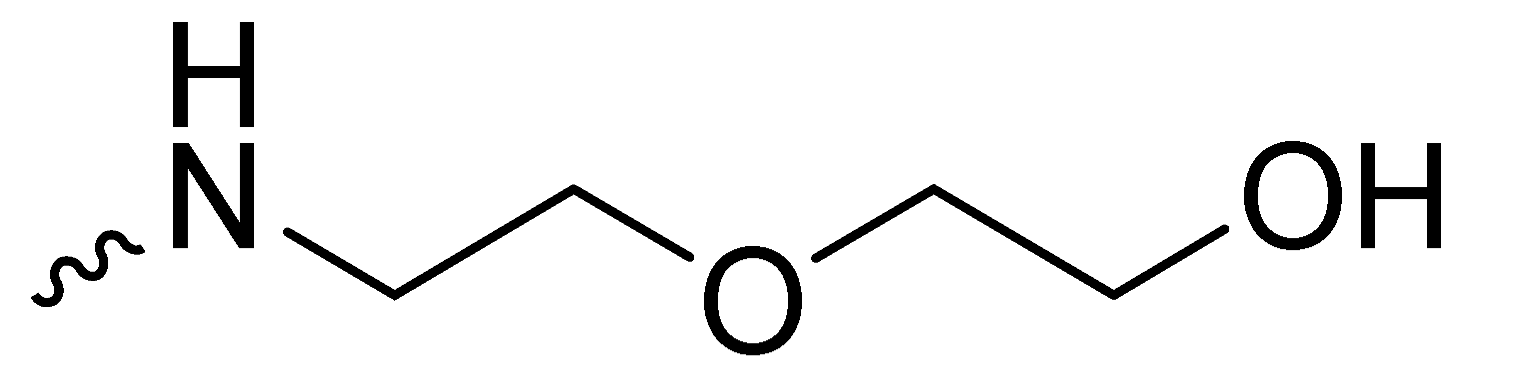 | 35.30 ± 2.08 |
| 1j | -OCH3 |  | 14.33 ± 2.08 |
| 2a | -H |  | 11.43 ± 1.50 |
| 2b | -OCH3 |  | 5.90 ± 0.26 |
| 2c | -H |  | 18.00 ± 4.58 |
| 2d | -OCH3 | 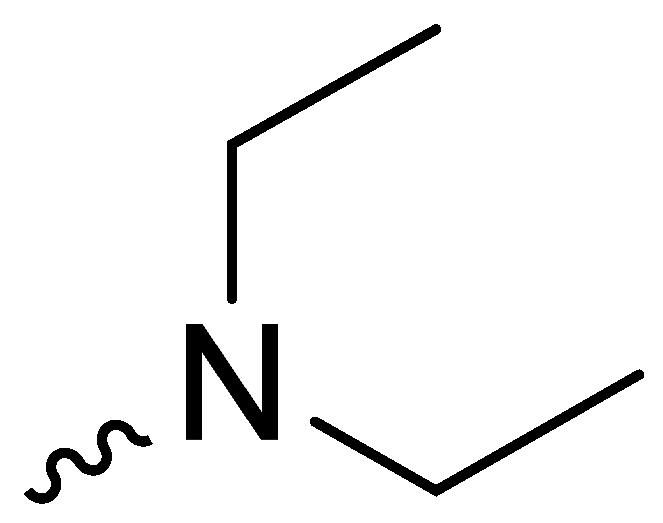 | 22.17 ± 1.75 |
| 2e | -H | 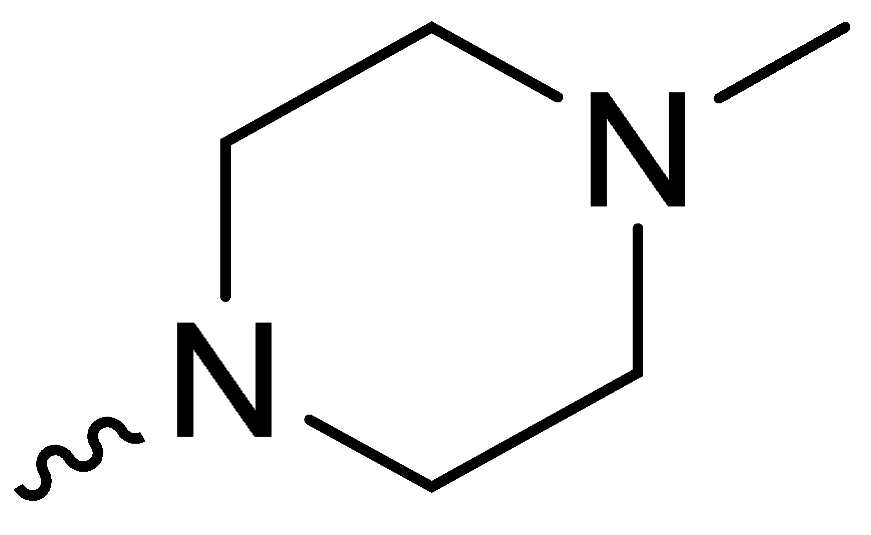 | 26.33 ± 2.08 |
| 2f | -OCH3 | 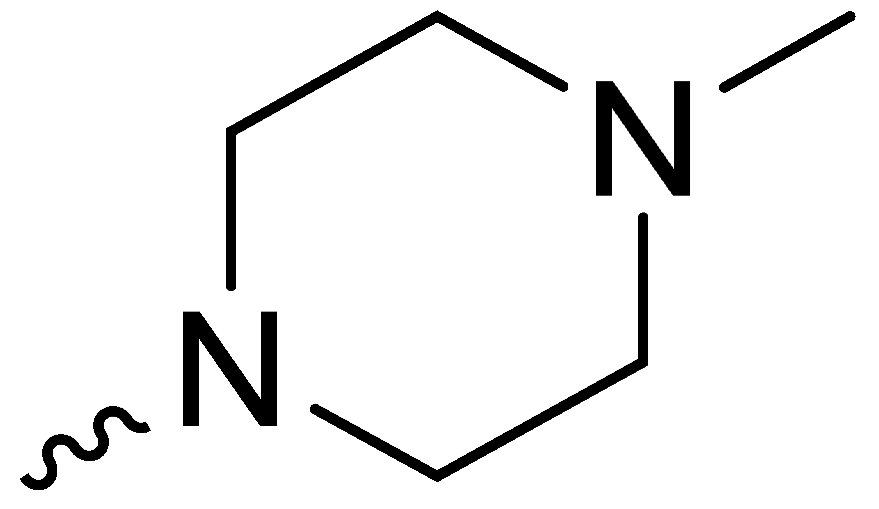 | 10.50 ± 0.50 |
| 2g | -H | 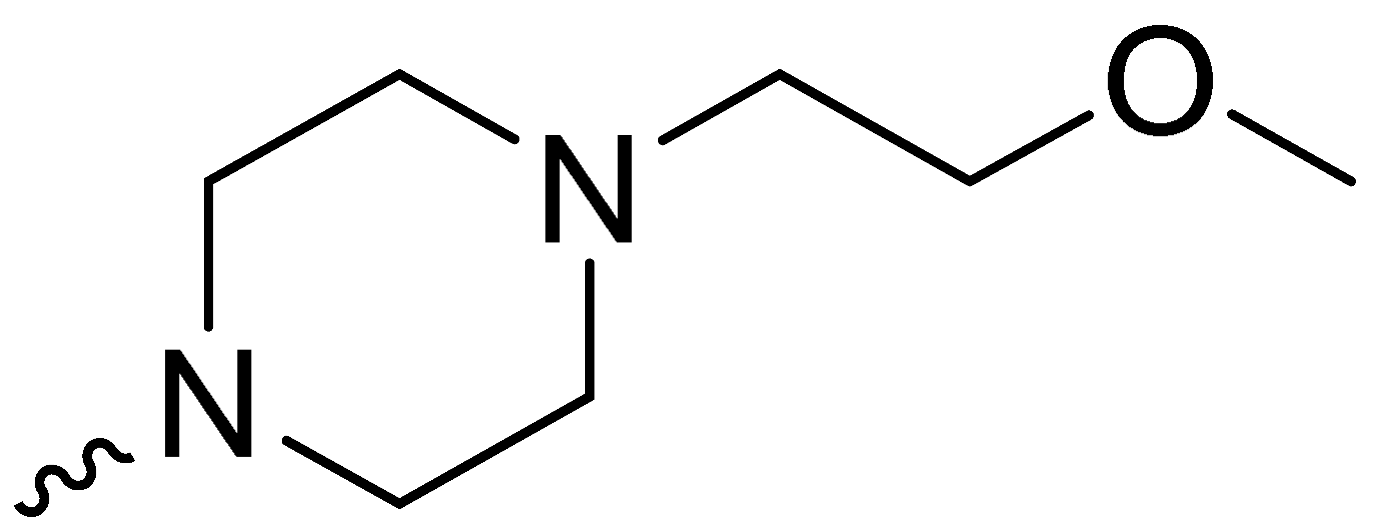 | 47.33 ± 24.09 |
| 2h | -OCH3 | 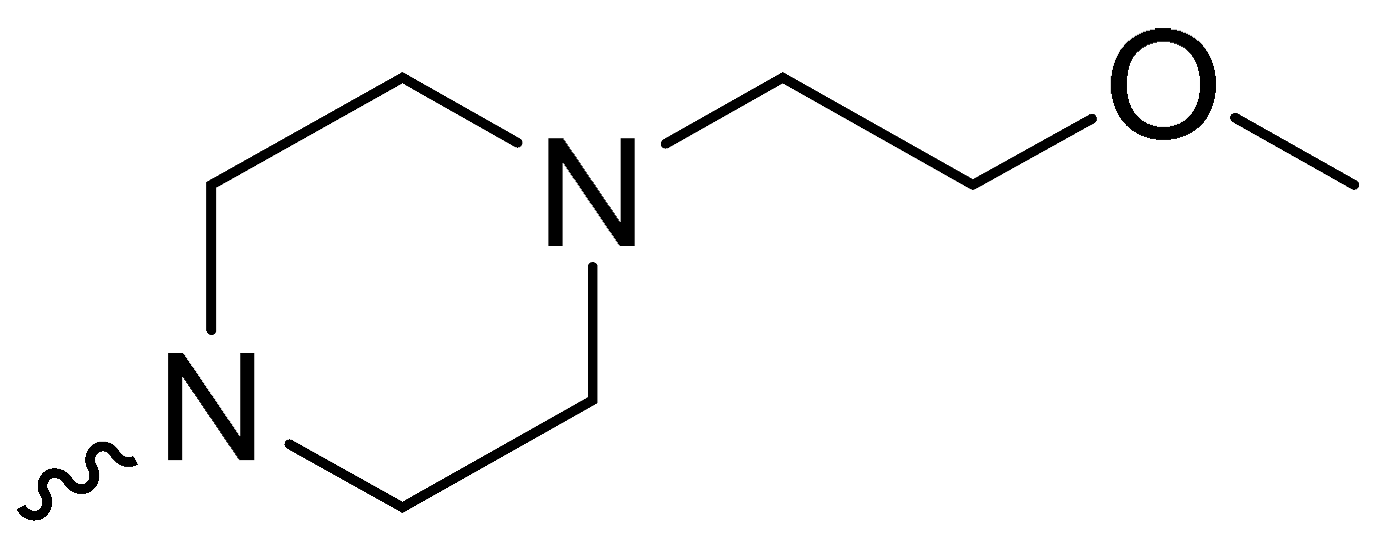 | 17.53 ± 1.28 |
| 2i | -H | 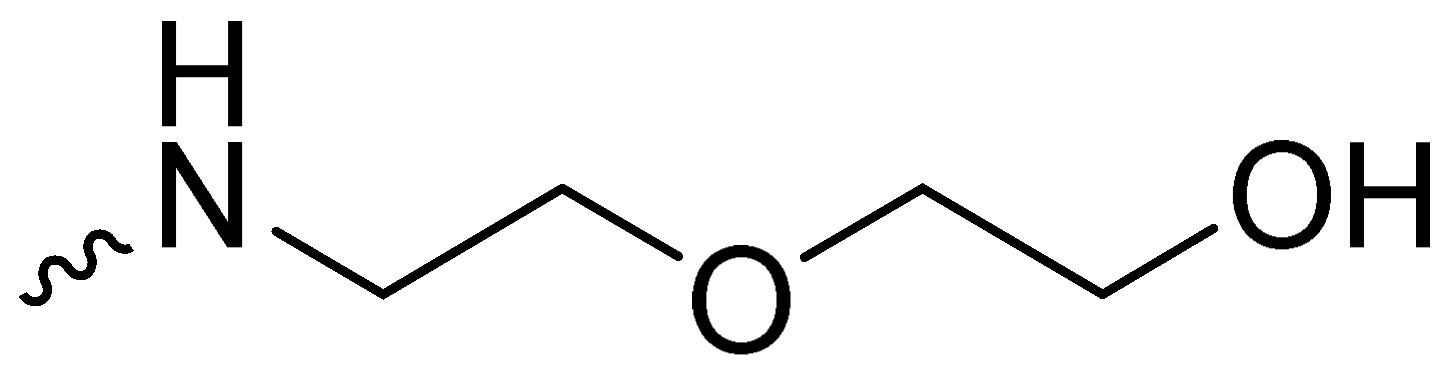 | 8.00 ± 1.00 |
| 2j | -OCH3 | 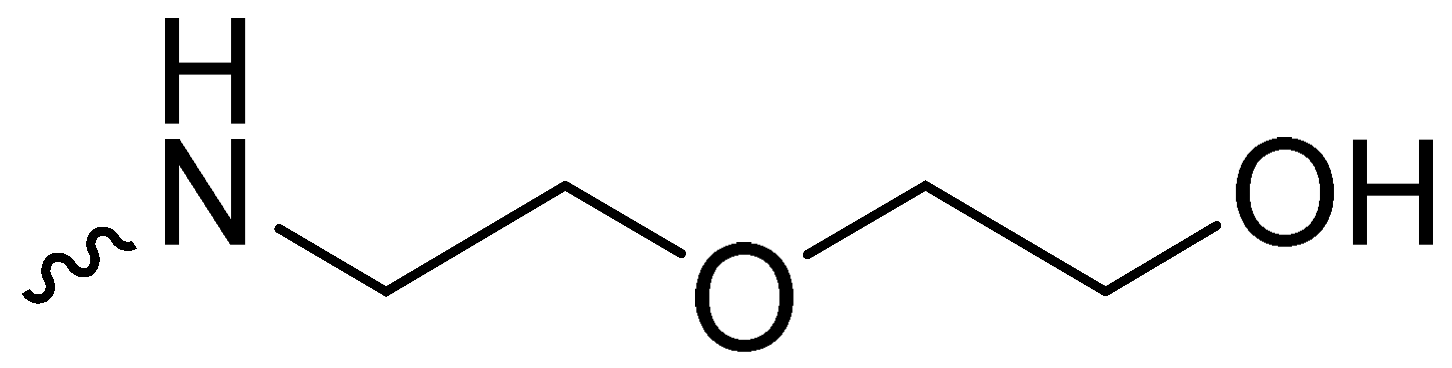 | 6.42 ± 0.24 |
| H1/H2/Me | Δδ c | H6/H8 | Δδ c | H1′ | |
|---|---|---|---|---|---|
| T4 | 1.57 | −0.09 | 7.09 | −0.12 | 5.72 |
| G5 | - | - | - | - | - |
| A6 | n.d. | - | 7.70 | +0.10 | n.d. |
| G7 | 11.36 | −0.38 | 7.83 | −0.19 | 6.08 |
| G8 | 10.98 | −0.24 | 7.68 | −0.04 | 6.31 |
| G9 | 10.56 | −0.04 | 7.78 | +0.03 | 6.27 |
| T10 | 2.00 | 0.00 | 7.87 | +0.06 | 6.53 |
| G11 | 11.14 | −0.56 | 7.83 | −0.16 | 6.02 |
| G12 | 11.14 | −0.35 | 7.73 | −0.17 | 6.10 |
| G13 | 10.67 | −0.38 | 7.74 | −0.10 | 6.41 |
| T14 | 1.95 | +0.03 | 7.68 | −0.03 | 6.25 |
| A15 | 8.36 | −0.01 | 8.56 | +0.04 | 6.66 |
| G16 | 11.48 | −0.42 | 7.97 | −0.14 | 6.31 |
| G17 | 11.04 | −0.20 | 7.66 | −0.14 | 6.10 |
| G18 | 10.69 | −0.32 | 7.80 | +0.01 | 6.42 |
| T19 | 2.00 | 0.00 | 7.88 | +0.02 | 6.53 |
| G20 | 11.19 | −0.09 | 8.03 | +0.14 | 6.00 |
| G21 | 11.19 | −0.16 | 7.81 | −0.10 | 6.02 |
| G22 | 10.76 | −0.27 | 7.81 | +0.20 | 6.02 |
| T23 | 1.80 | +0.32 | 7.09 | −0.05 | 5.62 |
| A24 | 7.43 | +0.35 | - | - | - |
| A25 | 7.47 | +0.10 | 7.57 | +0.08 | 5.90 |
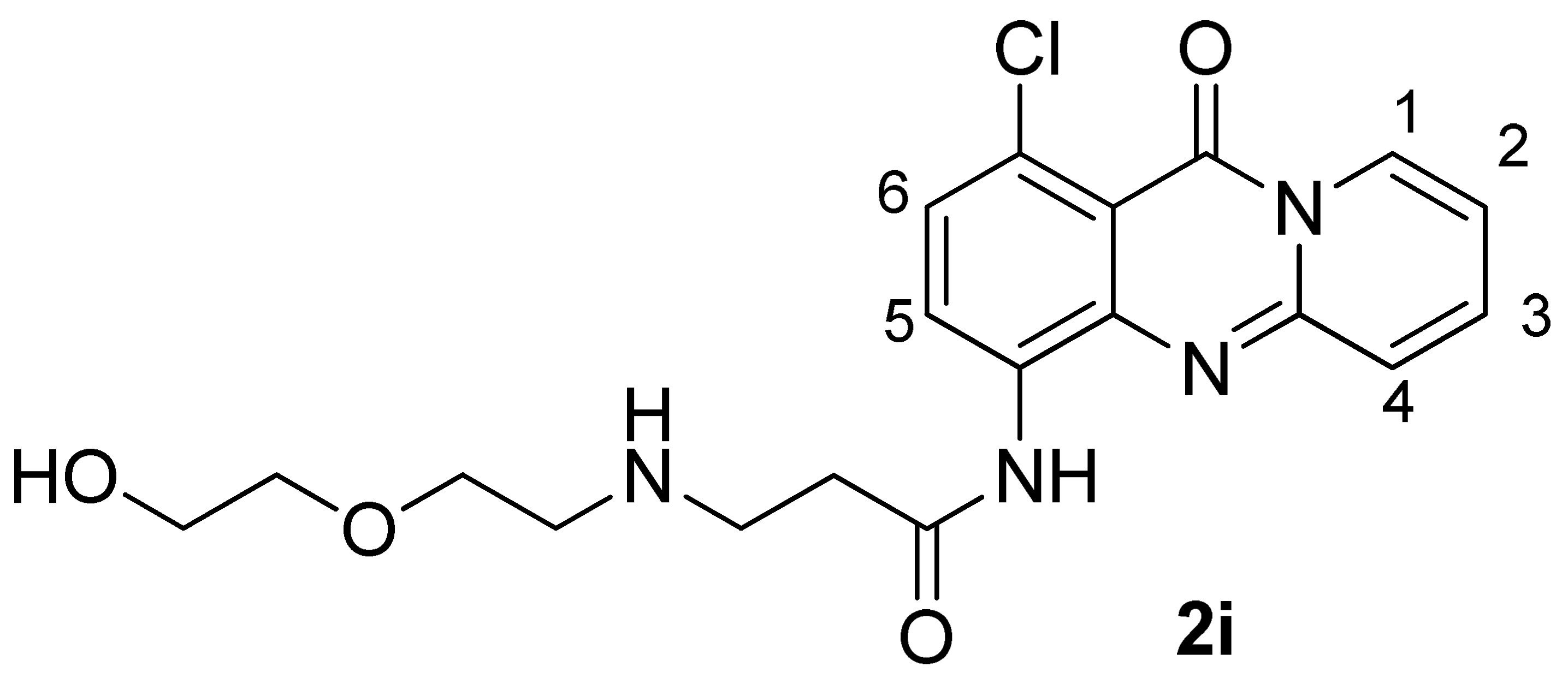
| 2i | Complex 2i/Pu22-T14T23 | Δδ b | |
|---|---|---|---|
| H1 | 8.65 | 8.23 | −0.42 |
| H2 | 7.13 | 6.62 | −0.51 |
| H3 | 7.78 | 7.26 | −0.52 |
| H4 | 7.57 | 6.91 | −0.66 |
| H5 | 8.23 | 7.70 | −0.53 |
| H6 | 7.35 | 6.66 | −0.69 |
| CH2CO | 2.57 | 2.70 | +0.13 |
| CH2NH | 3.11 | n.d. | - |
| CH2NH | 3.39 | n.d. | - |
| CH2O | 3.54 | 3.60 | +0.06 |
| CH2O | 3.65 | 3.78 | +0.13 |
| CH2OH | 3.82 | 3.88 | +0.06 |
| 2i | Pu22-T14T23 | |
|---|---|---|
| 5′-binding site | ||
| H2, H3, H4 | H8G7 | |
| H5 | NHG11 | |
| H5, H6 | H8G16 | |
| H3 | H1′G16 | |
| H5 | H1′G16 | |
| 3′-binding site | ||
| H4 | H1′G9 | |
| H4 | H8G22 | |
| H1 | H1′G22 | |
| H1, H2, H3, H4 | NHG18 | |
| H4 | NHG13 | |
| H3 | NHG22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Princiotto, S.; Karelou, M.; Ioannidi, R.; Beretta, G.L.; Zaffaroni, N.; Artali, R.; Kostakis, I.K.; Mazzini, S.; Dallavalle, S. Exploring the Interaction of New Pyridoquinazoline Derivatives with G-Quadruplex in the c-MYC Promoter Region. Int. J. Mol. Sci. 2023, 24, 14346. https://doi.org/10.3390/ijms241814346
Princiotto S, Karelou M, Ioannidi R, Beretta GL, Zaffaroni N, Artali R, Kostakis IK, Mazzini S, Dallavalle S. Exploring the Interaction of New Pyridoquinazoline Derivatives with G-Quadruplex in the c-MYC Promoter Region. International Journal of Molecular Sciences. 2023; 24(18):14346. https://doi.org/10.3390/ijms241814346
Chicago/Turabian StylePrinciotto, Salvatore, Maria Karelou, Rachel Ioannidi, Giovanni Luca Beretta, Nadia Zaffaroni, Roberto Artali, Ioannis K. Kostakis, Stefania Mazzini, and Sabrina Dallavalle. 2023. "Exploring the Interaction of New Pyridoquinazoline Derivatives with G-Quadruplex in the c-MYC Promoter Region" International Journal of Molecular Sciences 24, no. 18: 14346. https://doi.org/10.3390/ijms241814346
APA StylePrinciotto, S., Karelou, M., Ioannidi, R., Beretta, G. L., Zaffaroni, N., Artali, R., Kostakis, I. K., Mazzini, S., & Dallavalle, S. (2023). Exploring the Interaction of New Pyridoquinazoline Derivatives with G-Quadruplex in the c-MYC Promoter Region. International Journal of Molecular Sciences, 24(18), 14346. https://doi.org/10.3390/ijms241814346