

Toward a Unifying Hypothesis for Redesigned Lipid Catabolism as a Clinical Target in Advanced, Treatment-Resistant Carcinomas

Abstract
:1. Introduction
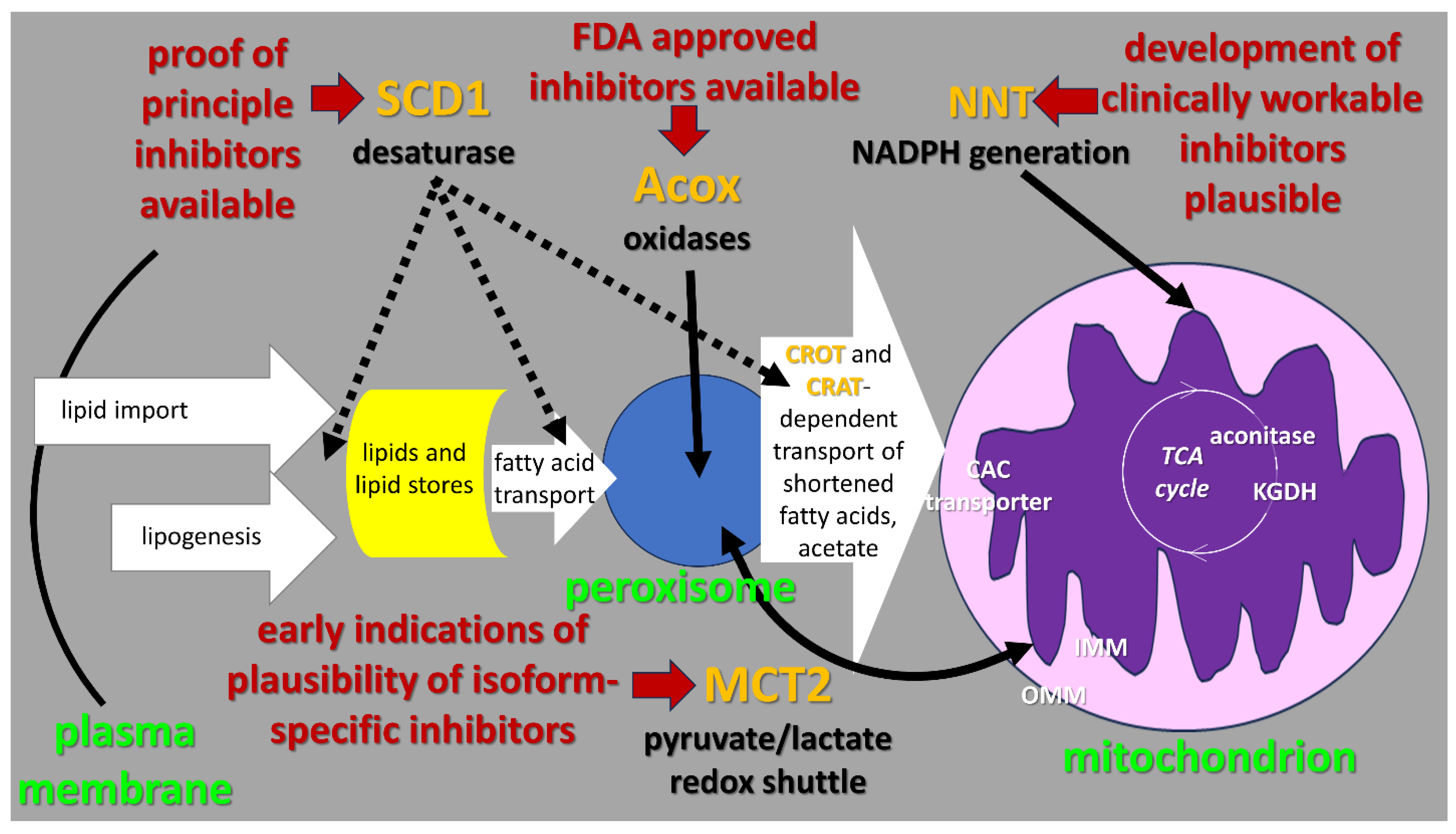
2. Context of “Lipid Metabolism Resistance System” (LMRS) Hypothesis
3. Detailed Description of the Steps in the LMRS Hypothesis
3.1. Mitochondrial Oxygen-Dependent Metabolism of Preprocessed Fatty Acids
3.2. Acox-Catalyzed, Oxygen-Dependent Peroxisomal Fatty Acid Beta-Oxidation
3.3. Oxygen-Dependent Fatty Acid Desaturation by the SCD1 Enzyme Anchored on the Cytoplasmic Face of the Endoplamic Reticulum (ER)
4. Conclusions and Practical Clinical Implications
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dvorak, H.F. Tumors—Wounds that do not heal—Similarities between tumor stroma generation and wound-healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Singer, A.J.; Clark, R.A.F. Mechanisms of disease—Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Riss, J.; Khanna, C.; Koo, S.; Chandramouli, G.V.R.; Yang, H.H.; Hu, Y.; Kleiner, D.E.; Rosenwald, A.; Schaefer, C.F.; Ben-Sasson, S.A.; et al. Cancers as wounds that do not heal: Differences and similarities between renal regeneration/repair and renal cell carcinoma. Cancer Res. 2006, 66, 7216–7224. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.S.; Gardner, K. Wounds that will not heal Pervasive cellular reprogramming in cancer. Am. J. Pathol. 2013, 182, 1055–1064. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal—A historical perspective with focus on the fundamental roles of increased vascular permeability and clotting. Semin. Thromb. Hemost. 2019, 45, 576–592. [Google Scholar] [CrossRef]
- Wong, A.Y.; Whited, J.L. Parallels between wound healing, epimorphic regeneration and solid tumors. Development 2020, 147, dev181636. [Google Scholar] [CrossRef]
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharm. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.Y.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Martin-Perez, M.; Urdiroz-Urricelqui, U.; Bigas, C.; Benitah, S.A. The role of lipids in cancer progression and metastasis. Cell Metab. 2022, 34, 1675–1699. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Zhao, F.P.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Kim, H.M.; Koo, J.S. Expression of lipid metabolism-related proteins in metastatic breast cancer. PLoS ONE 2015, 10, e0137204. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. A FATal combination: Fibroblast-derived lipids and cancer-derived autotaxin promote pancreatic cancer growth. Cancer Disc. 2019, 9, 578–580. [Google Scholar] [CrossRef]
- Cao, Y.H. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Investig. 2019, 129, 3006–3017. [Google Scholar] [CrossRef]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid metabolism and resistance to anticancer treatment. Biology 2020, 9, 474. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gasparovic, A.C.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Updates 2020, 49, 100670. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Dlubek, J.; Rysz, J.; Jablonowski, Z.; Gluba-Brzozka, A.; Franczyk, B. The correlation between lipid metabolism disorders and prostate cancer. Curr. Med. Chem. 2021, 28, 2048–2061. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, A.; Bellanger, D.; Guibon, R.; Bruyere, F.; Brisson, L.; Fromont, G. Lipophagy and prostate cancer: Association with disease aggressiveness and proximity to periprostatic adipose tissue. J. Path. 2021, 255, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef]
- Schiliro, C.; Firestein, B.L. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- An, D.D.; Zhai, D.Y.; Wan, C.; Yang, K.Y. The role of lipid metabolism in cancer radioresistance. Clin. Transl. Oncol. 2023, 25, 2332–2349. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.L.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Massari, M.S.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- Aloia, A.; Mullhaupt, D.; Chabbert, C.D.; Eberhart, T.; Fluckiger-Mangual, S.; Vukolic, A.; Eichhoff, O.; Irmisch, A.; Alexander, L.T.; Scibona, E. A fatty acid oxidation-dependent metabolic shift regulates the adaptation of BRAF-mutated melanoma to MAPK inhibitors. J. Clin. Investig. 2019, 25, 6852–6867. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers 2018, 10, 3. [Google Scholar] [CrossRef]
- Benzarti, M.; Delbrouck, C.; Neises, L.; Kiweler, N.; Meiser, J. Metabolic potential of cancer cells in context of the metastatic cascade. Cells 2020, 9, 2035. [Google Scholar] [CrossRef]
- Azam, A.; Sounni, N.E. Lipid metabolism heterogeneity and crosstalk with mitochondria functions drive breast cancer progression and drug resistance. Cancers 2022, 14, 6267. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.W.; Wang, B.L.; Chen, K.L.; Ding, R.; Wu, J.C.; Pan, Y.; Ji, P.; Ye, B.; Xiang, M.L. Perspectives of lipid metabolism reprogramming in head and neck squamous cell carcinoma: An overview. Front. Oncol. 2022, 12, 1008361. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.X.; Yi, M.; Xiang, B. Novel insights on lipid metabolism alterations in drug resistance in cancer. Front. Cell Dev. Biol. 2022, 10, 875318. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L. Fatty acid uptake and lipid storage induced by HIF-1 alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Updates 2011, 14, 191–201. [Google Scholar] [CrossRef]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef]
- Zhang, X.D.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.H.; Wang, L.G.; Ho, T.H.; Liu, J. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife 2017, 6, e31132. [Google Scholar] [CrossRef] [PubMed]
- Castelli, S.; Ciccarone, F.; Tavian, D.; Ciriolo, M.R. ROS-dependent HIF1 alpha activation under forced lipid catabolism entails glycolysis and mitophagy as mediators of higher proliferation rate in cervical cancer cells. J. Exp. Clin. Caner Res. 2021, 40, 94. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. AMPK-mTOR Signaling and cellular adaptations in hypoxia. Int. J. Mol. Sci. 2021, 22, 9765. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer cell metabolism in hypoxia: Role of HIF-1 as key regulator and therapeutic target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Yun, J.E.; Kim, S.J.; Chun, Y.S. Lipid metabolic reprogramming by hypoxia-inducible factor-1 in the hypoxic tumour microenvironment. Pflug. Arch. Eur. J. Physiol. 2022, 474, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Salavaty, A.; Azadian, E.; Naik, S.H.; Currie, P.D. Clonal selection parallels between normal and cancer tissues. Trend Genet. 2023, 39, 358–380. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Lasorda, F.; di Meo, N.A.; Rutigliano, M.; Ferro, M.; Terracciano, D.; Tataru, O.S.; Battaglia, M.; Ditonno, P.; Lucarelli, G. Emerging hallmarks of metabolic reprogramming in prostate cancer. Int. J. Mol. Sci. 2023, 24, 910. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, C.Y.; Martincuks, A.; Herrmann, A.; Yu, H. STAT proteins in cancer: Orchestration of metabolism. Nat. Rev. Cancer 2023, 23, 115–134. [Google Scholar] [CrossRef]
- Tadros, S.; Shukla, S.K.; King, R.J.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Yu, F.; Lazenby, A.J.; Berim, L.; et al. De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 2017, 77, 5503–5517. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase: A druggable driver of breast cancer brain metastasis. Expert Opin. Ther. Targets 2022, 26, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tao, J.Y.; Calvisi, D.F.; Chen, X. Role of lipogenesis rewiring in hepatocellular carcinoma. Sem. Liver Dis. 2022, 42, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Kinameri, A.; Suzuki, S.; Senga, S.; Ke, Y.Q.; Fujii, H. The cancer-promoting gene fatty acid-binding protein 5 (FABP5) is epigenetically regulated during human prostate carcinogenesis. Biochem. J. 2016, 473, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–49. [Google Scholar] [CrossRef]
- Cheng, C.M.; Geng, F.; Cheng, X.; Guo, D.L. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Su, F.; Ahn, S.; Saha, A.; DiGiovanni, J.; Kolonin, M.G. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene 2019, 38, 1979–1988. [Google Scholar] [CrossRef]
- Wang, J.C.; Li, Y.S. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498-U1207. [Google Scholar] [CrossRef]
- Singh, S.R.; Zeng, X.K.; Zhao, J.S.; Liu, Y.; Hou, G.; Liu, H.H.; Hou, S.X. The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature 2016, 538, 109–115. [Google Scholar] [CrossRef]
- Okumura, T.; Ohuchida, K.; Sada, M.; Abe, T.; Endo, S.; Koikawa, K.; Iwamoto, C.; Miura, D.; Mizuuchi, Y.; Moriyama, T.; et al. Extra-pancreatic invasion induces lipolytic and fibrotic changes in the adipose microenvironment, with released fatty acids enhancing the invasiveness of pancreatic cancer cells. Oncotarget 2017, 8, 18280–18295. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Attane, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, A.S.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef] [PubMed]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tat-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cell Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Lin, Y.Y.; Zhang, H.Q.; Liu, C.Q.; Cheng, Z.; Yang, X.W.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267. [Google Scholar] [CrossRef]
- Kay, E.J.; Zanivan, S. Metabolic pathways fuelling protumourigenic cancer-associated fibroblast functions. Curr. Opin. Syst. Biol. 2021, 28, 28. [Google Scholar] [CrossRef]
- Sugai, M.; Yanagawa, N.; Shikanai, S.; Hashimoto, M.; Saikawa, H.; Osakabe, M.; Saito, H.; Maemondo, M.; Sugai, T. Correlation of tumor microenvironment-related markers with clinical outcomes in patients with squamous cell carcinoma of the lung. Transl. Lung Cancer Res. 2022, 11, 975–985. [Google Scholar] [CrossRef]
- Andreucci, E.; Fioretto, B.S.; Rosa, I.; Matucci-Cerinic, M.; Biagioni, A.; Romano, E.; Calorini, L.; Manetti, M. Extracellular Lactic Acidosis of the Tumor Microenvironment Drives Adipocyte-to-Myofibroblast Transition Fueling the Generation of Cancer-Associated Fibroblasts. Cells 2023, 12, 939. [Google Scholar] [CrossRef]
- Hamabe-Horiike, T.; Harada, S.I.; Yoshida, K.; Kinoshita, J.; Yamaguchi, T.; Fushida, S. Adipocytes contribute to tumor progression and invasion of peritoneal metastasis by interacting with gastric cancer cells as cancer associated fibroblasts. Cancer Rep. 2023, 6, e1647. [Google Scholar] [CrossRef]
- Sato, S.; Hiruma, T.; Koizumi, M.; Yoshihara, M.; Nakamura, Y.; Tadokoro, H.; Motomatsu, S.; Yamanaka, T.; Washimi, K.; Okubo, Y.; et al. Bone marrow adipocytes induce cancer-associated fibroblasts and immune evasion, enhancing invasion and drug resistance. Cancer Sci. 2023, 114, 2674–2688. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. BBA Mol. Cell Biol. Lipids 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a matter of fat: The crosstalk between adipose tissue and tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; Song, E.W. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Sacca, P.A.; Calvo, J.C. Periprostatic adipose tissue microenvironment: Metabolic and hormonal pathways during prostate cancer progression. Front. Endocrinol. 2022, 13, 863027. [Google Scholar] [CrossRef]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-associated fibroblasts: The chief architect in the tumor microenvironment. Front. Cell Dev. Biol. 2023, 11, 1089068. [Google Scholar] [CrossRef]
- Sherman, M.H.; di Magliano, M.P. Cancer-associated fibroblasts: Lessons from pancreatic cancer. Annu. Rev. Cancer Biol. 2023, 7, 43–55. [Google Scholar] [CrossRef]
- Mailloux, R.J. Mitochondrial antioxidants and the maintenance of cellular hydrogen peroxide levels. Oxidative Med. Cell. Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. alpha-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Rad. Res. 2011, 45, 29–36. [Google Scholar] [CrossRef]
- Solmonson, A.; DeBerardinis, R.J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 2018, 293, 7522–7530. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Grayson, C.; Koufos, O. Regulation of mitochondrial hydrogen peroxide availability by protein S-glutathionylation. Cells 2023, 12, 107. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Palmieri, F.; Indiveri, C. The mitochondrial carnitine acyl-carnitine carrier (SLC25A20): Molecular mechanisms of transport, role in redox sensing and interaction with drugs. Biomolecules 2021, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Kisty, E.A.; Saart, E.C.; Weerapana, E. Identifying redox-sensitive cysteine residues in mitochondria. Antioxidants 2023, 12, 992. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2014, 19, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed]
- Castelli, S.; De Falco, P.; Ciccarone, F.; Desideri, E.; Ciriolo, M.R. Lipid catabolism and ROS in cancer: A bidirectional liaison. Cancers 2021, 13, 5484. [Google Scholar] [CrossRef]
- Castelli, S.; Ciccarone, F.; De Falco, P.; Ciriolo, M.R. Adaptive antioxidant response to mitochondrial fatty acid oxidation determines the proliferative outcome of cancer cells. Cancer Lett. 2023, 554, 216010. [Google Scholar] [CrossRef]
- Ying, M.F.; Hu, X. Tracing the electron flow in redox metabolism: The appropriate distribution of electrons is essential to maintain redox balance in cancer cells. Semin. Cancer Biol. 2022, 87, 32–47. [Google Scholar] [CrossRef]
- Kampjut, D.; Sazanov, L.A. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature 2019, 573, 291–295. [Google Scholar] [CrossRef]
- Jackson, J.B. A review of the binding-change mechanism for proton-translocating transhydrogenase. BBA Bioenerg. 2012, 1817, 1839–1846. [Google Scholar] [CrossRef]
- Nesci, S.; Trombetti, F.; Pagliarani, A. Nicotinamide nucleotide transhydrogenase as a sensor of mitochondrial biology. Trends Cell Biol. 2020, 30, 1–3. [Google Scholar] [CrossRef]
- Francisco, A.; Figueira, T.R.; Castilho, R.F. Mitochondrial NAD(P)(+) transhydrogenase: From molecular features to physiology and disease. Antioxid. Redox Signal. 2022, 36, 864–884. [Google Scholar] [CrossRef] [PubMed]
- Rydstrom, J. Site-specific inhibitors of mitochondrial nicotinamide-nucleotide transhydrogenase. Eur. J. Biochem. 1972, 31, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Bicego, R.; Francisco, A.; Ruas, J.S.; Siqueira-Santos, E.S.; Castilho, R.F. Undesirable effects of chemical inhibitors of NAD(P)(+) transhydrogenase on mitochondrial respiratory function. Arch. Biochem. Biophys. 2020, 692, 692. [Google Scholar] [CrossRef] [PubMed]
- Rydstrom, J. Mitochondrial NADPH, transhydrogenase and disease. BBA Bioenerg. 2006, 1757, 721–726. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, Y.Y.; Pan, Y.Q.; Zheng, X.J.; Liao, K.; Mo, H.Y.; Sheng, H.; Wu, Q.-N.; Liu, Z.-X.; Zeng, Z.-L.; et al. IL-10-associated NNT acetylation orchestrates iron-sulfur cluster maintenance and cancer immunotherapy resistance. Mol. Cell 2023, 83, 1887–1895. [Google Scholar] [CrossRef]
- Chortis, V.; Taylor, A.E.; Doig, C.L.; Walsh, M.D.; Meimaridou, E.; Jenkinson, C.; Rodriguez-Blanco, G.; Ronchi, C.L.; Jafri, A.; Metherell, L.A.; et al. Nicotinamide nucleotide transhydrogenase as a novel treatment target in adrenocortical carcinoma. Endocrinology 2018, 159, 2836–2849. [Google Scholar] [CrossRef]
- Gameiro, P.A.; Laviolette, L.A.; Kelleher, J.K.; Iliopoulos, O.; Stephanopoulos, G. Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J. Biol. Chem. 2013, 288, 12967–12977. [Google Scholar] [CrossRef]
- Li, S.; Zhuang, Z.N.; Wu, T.; Lin, J.C.; Liu, Z.X.; Zhou, L.F.; Dai, T.; Lu, L.; Ju, H.Q. Nicotinamide nucleotide transhydrogenase-mediated redox homeostasis promotes tumor growth and metastasis in gastric cancer. Redox Biol. 2018, 18, 246–255. [Google Scholar] [CrossRef]
- Ward, N.P.; Kang, Y.P.; Falzone, A.; Boyle, T.A.; DeNicola, G.M. Nicotinamide nucleotide transhydrogenase regulates mitochondrial metabolism in NSCLC through maintenance of Fe-S protein function. J. Exp. Med. 2020, 217, e20191689. [Google Scholar] [CrossRef]
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.J.; Song, Y.; Zou, X.J.; Liu, S.S.; Zhang, W.L.; Liu, L. Nicotinamide nucleotide transhydrogenase acts as a new prognosis biomarker in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2020, 13, 972–978. [Google Scholar] [PubMed]
- Lin, X.J.; Wu, W.Z.; Ying, Y.K.; Luo, J.; Xu, X.H.; Zheng, L.X.; Wu, W.; Yang, S.; Zhao, S.K. MicroRNA-31: A pivotal oncogenic factor in oral squamous cell carcinoma. Cell Death Discov. 2022, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Henne, W.M.; Reese, M.L.; Goodman, J.M. The assembly of lipid droplets and their roles in challenged cells. EMBO J. 2018, 37, e98947. [Google Scholar] [CrossRef]
- Klemm, R.W. Getting in touch is an important step: Control of metabolism at organelle contact sites. Sage J. 2021, 4, 2515256421993708. [Google Scholar] [CrossRef]
- Renne, M.F.; Hariri, H. Lipid droplet-organelle contact sites as hubs for fatty acid metabolism, trafficking, and metabolic channeling. Front. Cell Dev. Biol. 2021, 9, 726261. [Google Scholar] [CrossRef]
- Kim, J.A. Peroxisome metabolism in cancer. Cells 2020, 9, 1692. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Di Pietro, E.; Jangal, M.; Goncalves, C.; Witcher, M.; Braverman, N.E.; del Rincon, S.V. Peroxisomes and cancer: The role of a metabolic specialist in a disease of aberrant metabolism. BBA Rev. Cancer 2018, 1870, 103–121. [Google Scholar] [CrossRef]
- Ravi, A.; Palamiuc, L.; Emerling, B.M. Crucial players for inter-organelle communication: PI5P4Ks and their lipid product PI-4,5-P-2 come to the surface. Front. Cell Dev. Biol. 2022, 9, 791758. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated fat oxidation, mechanisms, and therapeutic potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J.A. Human disorders of peroxisome metabolism and biogenesis. BBA Mol. Cell Res. 2016, 1863, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.J.; Sun, X.; Wang, W.C.; Lian, Z.S.; Wu, P.; Han, S.X.; Chen, H.; Zhang, P.M. Disruption of peroxisome function leads to metabolic stress, mTOR inhibition, and lethality in liver cancer cells. Caner Lett. 2018, 421, 82–93. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: A metabolic survival role for tumor-associated stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef]
- Valenca, I.; Pertega-Gomes, N.; Vizcaino, J.R.; Henrique, R.M.; Lopes, C.; Baltazar, F.; Ribeiro, D. Localization of MCT2 at peroxisomes is associated with malignant transformation in prostate cancer. J. Cell. Mol. Med. 2015, 19, 723–733. [Google Scholar] [CrossRef]
- Valenca, I.; Ferreira, A.R.; Correia, M.; Kuhl, S.; van Roermund, C.; Waterham, H.R.; Maximo, V.; Islinger, M.; Ribeiro, D. Prostate cancer proliferation is affected by the subcellular localization of MCT2 and accompanied by significant peroxisomal alterations. Cancers 2020, 12, 3152. [Google Scholar] [CrossRef]
- Kane, D.A. Lactate oxidation at the mitochondria: A lactate-malate-aspartate shuttle at work. Front. Neurosci. 2014, 8, 366. [Google Scholar] [CrossRef]
- Shen, S.S.; Faouzi, S.; Souquere, S.; Roy, S.; Routier, E.; Libenciuc, C.; Andre, F.; Pierron, G.; Scoazec, J.-Y.; Robert, C. Melanoma persister cells are tolerant to BRAF/MEK inhibitors via ACOX1-mediated fatty acid oxidation. Cell Rep. 2020, 33, 108421. [Google Scholar] [CrossRef]
- Lasheras-Otero, I.; Feliu, I.; Maillo, A.; Moreno, H.; Redondo-Munoz, M.; Aldaz, P.; Bocanegra, A.; Olias-Arjona, A.; Lecanda, F.; Fernandez-Irigoyen, J.; et al. The regulators of peroxisomal acyl-carnitine shuttle CROT and CRAT promote metastasis in melanoma. J. Investig. Dermatol. 2023, 143, 305–318. [Google Scholar] [CrossRef]
- Guardado Rivas, M.O.; Stuart, S.D.; Thach, D.; Dahan, M.; Shorr, R.; Zachar, Z.; Bingham, P.M. Evidence for a novel, effective approach to targeting carcinoma catabolism exploiting the first-in-class, anti-cancer mitochondrial drug, CPI-613. PLoS ONE 2022, 17, e0269620. [Google Scholar] [CrossRef] [PubMed]
- Lauer, C.; Volkl, A.; Riedl, S.; Fahimi, H.D.; Beier, K. Impairment of peroxisomal biogenesis in human colon carcinoma. Carcinogenesis 1999, 20, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Frederiks, W.M.; Bosch, K.S.; Hoeben, K.A.; van Marle, J.; Langbein, S. Renal cell carcinoma and oxidative stress: The lack of peroxisomes. Acta Histochem. 2010, 112, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.S.; Ha, Z.Y.; Di Pietro, E.; Nichol, J.N.; Bolt, A.M.; Goncalves, C.; Dupere-Richer, D.; Pettersson, F.; Mann, K.K.; Braverman, N.E.; et al. Peroxisomes protect lymphoma cells from HDAC inhibitor-mediated apoptosis. Cell Death Differ. 2017, 24, 1912–1924. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.S.; Huang, F.; Goncalves, C.; Gonzalez, R.E.F.; Prabhu, S.; Bolt, A.; Di Pietro, E.; Khoury, E.; Heath, J.; Xu, Z.Y.; et al. Silencing PEX26 as an unconventional mode to kill drug-resistant cancer cells and forestall drug resistance. Autophagy 2022, 18, 540–558. [Google Scholar] [CrossRef]
- Zheng, F.M.; Chen, W.B.; Qin, T.; Lv, L.N.; Feng, B.; Lu, Y.L.; Li, Z.-Q.; Wang, X.-C.; Tao, L.-J.; Li, H.-W.; et al. ACOX1 destabilizes p73 to suppress intrinsic apoptosis pathway and regulates sensitivity to doxorubicin in lymphoma cells. BMB Rep. 2019, 52, 566–571. [Google Scholar] [CrossRef]
- Kim, S.; Lee, Y.; Koo, J.S. Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PLoS ONE 2015, 10, e0119473. [Google Scholar] [CrossRef]
- Yu, G.Y.; Cheng, C.J.; Lin, S.C.; Lee, Y.C.; Frigo, D.E.; Yu-Lee, L.Y.; Gallick, G.E.; Titus, M.A.; Nutt, L.K.; Lin, S.H. Organelle-derived acetyl-CoA promotes prostate cancer cell survival, migration, and metastasis via activation of calmodulin kinase II. Cancer Res. 2018, 78, 2490–2502. [Google Scholar] [CrossRef]
- Kuna, R.S.; Kumar, A.; Wessendorf-Rodriguez, K.A.; Galvez, H.; Green, C.R.; McGregor, G.H.; Cordes, T.; Shaw, R.J.; Svensson, R.U.; Metallo, C.M. Inter-organelle cross-talk supports acetyl-coenzyme A homeostasis and lipogenesis under metabolic stress. Sci. Adv. 2023, 9, eadf0138. [Google Scholar] [CrossRef]
- Tamatani, T.; Hattori, K.; Nakashiro, K.I.; Hayashi, Y.; Wu, S.Q.; Klumpp, D.; Reddy, J.K.; Oyasu, R. Neoplastic conversion of human urothelial cells in vitro by overexpression of H2O2-generating peroxisomal fatty acyl CoA oxidase. Int. J. Oncol. 1999, 15, 743–749. [Google Scholar] [CrossRef]
- Okamoto, M.; Reddy, J.K.; Oyasu, R. Tumorigenic conversion of a non-tumorigenic rat urothelial cell line by overexpression of H2O2-generating peroxisomal fatty acyl-CoA oxidase. Int. J. Cancer 1997, 70, 716–721. [Google Scholar] [CrossRef]
- Sellin, J.; Wingen, C.; Gosejacob, D.; Senyilmaz, D.; Hanschke, L.; Buttner, S.; Meyer, K.; Bano, D.; Nicotera, P.; Teleman, A.A.; et al. Dietary rescue of lipotoxicity-induced mitochondrial damage in Peroxin19 mutants. PLoS Biol. 2018, 16, e2004893. [Google Scholar] [CrossRef]
- Ben-David, U.; Gan, Q.F.; Golan-Lev, T.; Arora, P.; Yanuka, O.; Oren, Y.S.; Leikin-Frenkel, A.; Graf, M.; Garippa, R.; Boehringer, M.; et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell 2013, 12, 167–179. [Google Scholar] [CrossRef]
- Mohammadzadeh, F.H.V.; Alihemmati, A.; Shaaker, M.; Mosayyeb, G.; Darabi, M.; Mehdizadeh, A. The role of stearoyl-coenzyme A desaturase 1 in liver development, function, and pathogenesis. J. Ren. Hepatic Disord. 2019, 3, 15–22. [Google Scholar] [CrossRef]
- Xue, Y.Q.; Lin, L.Y.; Li, Q.; Liu, K.L.; Hu, M.Y.; Ye, J.Y.; Cao, J.; Zhai, J.; Zheng, F.; Wang, Y.; et al. SCD1 sustains homeostasis of bulge niche via maintaining hemidesmosomes in basal keratinocytes. Adv. Sci. 2023, 10, 2201949. [Google Scholar] [CrossRef] [PubMed]
- Aljohani, A.M.; Syed, D.N.; Ntambi, J.M. Insights into stearoyl-coa desaturase-1 regulation of systemic metabolism. Trends Endocrinol. Metab. 2017, 28, 831–842. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef]
- Igal, R.A. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. BBA Mol. Cell Biol. Lipids 2016, 1861, 1865–1880. [Google Scholar] [CrossRef]
- Ascenzi, F.; De Vitis, C.; Maugeri-Sacca, M.; Napoli, C.; Ciliberto, G.; Mancini, R. SCD1, autophagy and cancer: Implications for therapy. J. Exp. Clin. Cancer Res. 2021, 40, 265. [Google Scholar] [CrossRef]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.P.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [CrossRef]
- Peck, B.; Schug, Z.T.; Zhang, Q.; Dankworth, B.; Jones, D.T.; Smethurst, E.; Patel, R.; Mason, S.; Jiang, M.; Saunders, R.; et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab. 2016, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Tsukamoto, H. Stearoyl-CoA desaturase and tumorigenesis. Chem. Biol. Interact. 2020, 316, 108917. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.R.; Butler, L.M.; Hoy, A.J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 2021, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Kubota, C.S.; Espenshade, P.J. Targeting stearoyl-CoA desaturase in solid tumors. Cancer Res. 2022, 82, 1682–1688. [Google Scholar] [CrossRef]
- Raeisi, M.; Hassanbeigi, L.; Khalili, F.; Kharrati-Shishavan, H.; Yousefi, M.; Mehdizadeh, A. Stearoyl-CoA desaturase 1 as a therapeutic target for cancer: A focus on hepatocellular carcinoma. Mol. Biol. Rep. 2022, 49, 8871–8882. [Google Scholar] [CrossRef]
- Min, J.Y.; Kim, D. Stearoyl-CoA desaturase 1 as a therapeutic biomarker: Focusing on cancer stem cells. Int. J. Mol. Sci. 2023, 24, 8951. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, N.; Igal, R.A. Stearoyl-CoA desaturase is involved in the control of proliferation, anchorage-independent growth, and survival in human transformed cells. J. Biol. Chem. 2005, 280, 25339–25349. [Google Scholar] [CrossRef]
- Scaglia, N.; Chisholm, J.W.; Igal, R.A. Inhibition of stearoylCoA desaturase-1 inactivates acetyl-CoA carboxylase and impairs proliferation in cancer cells: Role of AMPK. PLoS ONE 2009, 4, e6812. [Google Scholar] [CrossRef]
- Xuan, Y.; Wang, H.G.; Yung, M.M.H.; Chen, F.S.; Chan, W.S.; Chan, Y.S.; Tsui, S.K.W.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. SCD1/FADS2 fatty acid desaturases equipoise lipid metabolic activity and redox-driven ferroptosis in ascites-derived ovarian cancer cells. Theranostics 2022, 12, 3534–3552. [Google Scholar] [CrossRef]
- Huang, J.; Fan, X.X.; He, J.X.; Pan, H.; Li, R.Z.; Huang, L.Y.; Jiang, Z.; Yao, X.-J.; Liu, L.; Leung, E.L.-H.; et al. SCD1 is associated with tumor promotion, late stage and poor survival in lung adenocarcinoma. Oncotarget 2016, 7, 39970–39979. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Noto, A.; De Vitis, C.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Caner Lett. 2017, 406, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Pisanu, M.E.; Maugeri-Sacca, M.; Fattore, L.; Bruschini, S.; De Vitis, C.; Tabbi, E.; Bellei, B.; Migliano, E.; Kovacs, D.; Camera, E.; et al. Inhibition of stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 318. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Yang, Y.; Jung, J.H.; Kim, Y. Oleic acid from cancer-associated fibroblast promotes cancer cell stemness by stearoyl-CoA desaturase under glucose-deficient condition. Cancer Cell Int. 2022, 22, 404. [Google Scholar] [CrossRef]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avances, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef]
- Budhu, A.; Roessler, S.; Zhao, X.L.; Yu, Z.P.; Forgues, M.; Ji, J.F.; Karoly, E.; Qin, L.-X.; Ye, Q.-H.; Jia, H.-L.; et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 2013, 144, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.K.F.; Lau, E.Y.T.; Leung, D.H.W.; Lo, J.; Ho, N.P.Y.; Cheng, L.K.W.; Ma, S.; Lin, C.H.; Copland, J.A.; Ding, J.; et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J. Hepatol. 2017, 67, 979–990. [Google Scholar] [CrossRef]
- von Roemeling, C.A.; Marlow, L.A.; Pinkerton, A.B.; Crist, A.; Miller, J.; Tun, H.W.; Smallridge, R.C.; Copland, J.A. Aberrant lipid metabolism in anaplastic thyroid carcinoma reveals stearoyl CoA desaturase 1 as a novel therapeutic target. J. Clin. Endocrinol. Metab. 2015, 100, E697–E709. [Google Scholar] [CrossRef]
- Liu, G.H.; Feng, S.; Jia, L.; Wang, C.Y.; Fu, Y.; Luo, Y.Z. Lung fibroblasts promote metastatic colonization through upregulation of stearoyl-CoA desaturase 1 in tumor cells. Oncogene 2018, 37, 1519–1533. [Google Scholar] [CrossRef]
- Noto, A.; Raffa, S.; De Vitis, C.; Roscilli, G.; Malpicci, D.; Coluccia, P.; Di Napoli, A.; Ricci, A.; Giovagnoli, M.R.; Aurisicchio, L.; et al. Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells. Cell Death Dis. 2013, 4, e947. [Google Scholar] [CrossRef]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef]
- Ran, H.; Zhu, Y.M.; Deng, R.Y.; Zhang, Q.; Liu, X.S.; Feng, M.; Zhong, J.; Lin, S.; Tong, X.; Su, Q. Stearoyl-CoA desaturase-1 promotes colorectal cancer metastasis in response to glucose by suppressing PTEN. J. Exp. Clin. Cancer Res. 2018, 37, 54. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Condello, S.; Thomes-Pepin, J.; Ma, X.X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 2017, 20, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Berk, M.; Alkhouri, N.; Partrick, D.A.; Fung, J.J.; Feldstein, A. Stearoyl-CoA desaturase plays an important role in proliferation and chemoresistance in human hepatocellular carcinoma. J. Surg. Res. 2014, 186, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, X.H.; Song, S.; Wang, Y.H.; Dan, Q.F.; Ge, H. Targeting stearoyl-CoA desaturase enhances radiation induced ferroptosis and immunogenic cell death in esophageal squamous cell carcinoma. Oncoimmunology 2022, 11, 2101769. [Google Scholar] [CrossRef]
- Zhang, R.H.; Liu, L.G.; Wang, F.Q.; Zhao, W.Z.; Liu, K.; Yu, H.; Zhao, S.; Xu, B.; Zhang, X.; Chai, J.; et al. AKAP8L enhances the stemness and chemoresistance of gastric cancer cells by stabilizing SCD1 mRNA. Cell Death Dis. 2022, 13, 1041. [Google Scholar] [CrossRef] [PubMed]
- Morais, C.M.; Cardoso, A.M.; Araujo, A.R.D.; Reis, A.; Domingues, P.; Domingues, M.R.M.; de Lima, M.C.P.; Jurado, A.S. Stearoyl CoA desaturase-1 silencing in glioblastoma cells: Phospholipid remodeling and cytotoxicity enhanced upon autophagy inhibition. Int. J. Mol. Sci. 2022, 23, 13014. [Google Scholar] [CrossRef]
- Parik, S.; Fernandez-Garcia, J.; Lodi, F.; De Vlaminck, K.; Derweduwe, M.; De Vleeschouwer, S.; Sciot, R.; Geens, W.; Weng, L.; Bosisio, F.M.; et al. GBM tumors are heterogeneous in their fatty acid metabolism and modulating fatty acid metabolism sensitizes cancer cells derived from recurring GBM tumors to temozolomide. Front. Oncol. 2022, 12, 988872. [Google Scholar] [CrossRef]
- Lien, E.C.; Westermark, A.M.; Zhang, Y.; Yuan, C.; Li, Z.Q.; Lau, A.N.; Sapp, K.M.; Wolpin, B.M.; Vander Heiden, M.G. (Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 2021, 599, 302–307. [Google Scholar] [CrossRef]
- Sun, M.; Chen, X.X.; Yang, Z.B. Single cell mass spectrometry studies reveal metabolomic features and potential mechanisms of drug-resistant cancer cell lines. Anal. Chim. Acta 2022, 1206, 339761. [Google Scholar] [CrossRef]
- Li, W.; Bai, H.; Liu, S.; Cao, D.; Wu, H.; Shen, K.; Tai, Y.; Yang, J. Targeting stearoyl-CoA desaturase 1 to repress endometrial cancer progression. Oncotarget 2018, 9, 12064–12078. [Google Scholar] [CrossRef]
- Piao, C.Y.; Cui, X.L.; Zhan, B.; Li, J.; Li, Z.L.; Li, Z.H.; Liu, X.; Bi, J.; Zhang, Z.; Kong, C.Z. Inhibition of stearoyl CoA desaturase-1 activity suppresses tumour progression and improves prognosis in human bladder cancer. J. Cell. Mol. Med. 2019, 23, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Xu, Y.Z.; Zhe, L.S.; Zou, Y.; Kong, W.; Dong, B.J.; Huang, J.; Chen, Y.; Xue, W.; Zhang, J. High expression of stearoyl-CoA desaturase 1 predicts poor prognosis in patients with clear cell renal cell carcinoma. PLoS ONE 2016, 11, e0166231. [Google Scholar] [CrossRef] [PubMed]
- Ducheix, S.; Peres, C.; Hardfeldt, J.; Frau, C.; Mocciaro, G.; Piccinin, E.; Lobaccaro, J.-M.; De Santis, S.; Chieppa, M.; Bertrand-Michel, J.; et al. Deletion of stearoyl-CoA desaturase-1 from the intestinal epithelium promotes inflammation and tumorigenesis, reversed by dietary oleate. Gastroenterology 2018, 155, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Gunes, B.A.; Hekmatshoar, Y.; Ozkan, T.; Bozkurt, S.; Aydos, O.S.E.; Buyukasik, Y.; Aladag, E.; Sunguroglu, A. Downregulation of stearoyl-CoA desaturase 1 (SCD-1) promotes resistance to imatinib in chronic myeloid leukemia. Mediterr. J. Hematol. Infect. Dis. 2023, 15, e2023008. [Google Scholar] [CrossRef]
- Van den Branden, C.; Roels, F. Thioridazine: A selective inhibitor of peroxisomal beta-oxidation in vivo. FEBS Lett. 1985, 187, 331–333. [Google Scholar] [CrossRef]
- Shi, R.L.; Zhang, Y.; Shi, Y.; Shi, S.Y.; Jiang, L.L. Inhibition of peroxisomal beta-oxidation by thioridazine increases the amount of VLCFAs and A beta generation in the rat brain. Neurosci. Lett. 2012, 528, 6–10. [Google Scholar] [CrossRef]
- Beach, S.R.; Celano, C.M.; Noseworthy, P.A.; Januzzi, J.L.; Huffman, J.C. QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics 2013, 54, 1–13. [Google Scholar] [CrossRef]
- Huang, X.; Gan, G.M.; Wang, X.X.; Xu, T.; Xie, W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy 2019, 15, 1258–1279. [Google Scholar] [CrossRef]
- Yu, D.L.; Li, Y.T.; Sun, K.D.Y.; Gu, J.J.; Chen, Z.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.Y. The novel MET inhibitor, HQP8361, possesses single agent activity and enhances therapeutic efficacy of AZD9291 (osimertinib) against AZD9291-resistant NSCLC cells with activated MET. Am. J. Cancer Res. 2020, 10, 3316–3327. [Google Scholar]
- Luttich, L.; Besso, M.J.; Heiden, S.; Koi, L.; Baumann, M.; Krause, M.; Dubrovska, A.; Linge, A.; Kurth, I.; Peitzsch, C. Tyrosine kinase c-MET as therapeutic target for radiosensitization of head and neck squamous cell carcinomas. Cancers 2021, 13, 1865. [Google Scholar] [CrossRef]
- Oh, H.N.; Kwak, A.W.; Lee, M.H.; Kim, E.; Yoon, G.; Cho, S.S.; Liu, K.; Chae, J.-I.; Shim, J.H. Targeted inhibition of c-MET by podophyllotoxin promotes caspase-dependent apoptosis and suppresses cell growth in gefitinib-resistant non-small cell lung cancer cells. Phytomedicine 2021, 80, 153355. [Google Scholar] [CrossRef] [PubMed]
- Terlecka, P.; Krawczyk, P.; Grenda, A.; Milanowski, J. MET gene dysregulation as a promising therapeutic target in lung cancer—A review. J. Pers. Med. 2021, 11, 1370. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Peng, X.D.; Qian, X.J.; Zhang, K.M.; Huang, X.; Chen, Y.H.; Li, Y.-T.; Feng, G.-K.; Zhang, H.-L.; Xu, X.-L.; et al. Fis1 phosphorylation by Met promotes mitochondrial fission and hepatocellular carcinoma metastasis. Signal Transduct. Target. Ther. 2021, 6, 401. [Google Scholar] [CrossRef]
- Chen, Z.; Vallega, K.A.; Chen, H.Y.; Zhou, J.; Ramalingam, S.S.; Sun, S.Y. The natural product berberine synergizes with osimertinib preferentially against MET-amplified osimertinib-resistant lung cancer via direct MET inhibition. Pharmacol. Res. 2022, 175, 105998. [Google Scholar] [CrossRef]
- Takumi, Y.; Arai, S.; Suzuki, C.; Fukuda, K.; Nishiyama, A.; Takeuchi, S.; Sato, H.; Matsumoto, K.; Sugio, K.; Yano, S. MET kinase inhibitor reverses resistance to entrectinib induced by hepatocyte growth factor in tumors with NTRK1 or ROS1 rearrangements. Cancer Med. 2023, 12, 5809–5820. [Google Scholar] [CrossRef]
- Xu, X.M.; Yao, L. Recent patents on the development of c-MET kinase inhibitors. Recent Pat. Anti Cancer Drug Discov. 2020, 15, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.T.; Zhang, J.Q.; Fu, D.X.; Liu, M.; Zhang, H.; Tang, S.; Wang, L.; Xu, S.; Zhu, W.; Tang, Q.; et al. Design, synthesis and biological evaluation of 4-(4-aminophenoxy) picolinamide derivatives as potential antitumor agents. Eur. J. Med. Chem. 2023, 257, 115499. [Google Scholar] [CrossRef]
- Michaelides, I.N.; Collie, G.W.; Borjesson, U.; Vasalou, C.; Alkhatib, O.; Barlind, L.; Cheung, T.; Dale, I.L.; Embrey, K.J.; Hennessy, E.J.; et al. Discovery and optimization of the first ATP competitive type-III c-MET inhibitor. J. Med. Chem. 2023, 13, 8782–8807. [Google Scholar] [CrossRef]
- Zeng, X.C.; Hu, Y.W.; Xu, Z.; Wu, X.Y.; Xiong, Y.; Liu, S.P. Development of new c-MET inhibitors and application of corresponding multitarget tyrosine kinase inhibitors in tumor therapy. Soft Comput. 2023; early access. [Google Scholar] [CrossRef]
- Ovens, M.J.; Manoharan, C.; Wilson, M.C.; Murray, C.M.; Halestrap, A.P. The inhibition of monocarboxylate transporter 2 (MCT2) by AR-C155858 is modulated by the associated ancillary protein. Biochem. J. 2010, 431, 217–225. [Google Scholar] [CrossRef]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef]
- Singh, M.; Afonso, J.; Sharma, D.; Gupta, R.; Kumar, V.; Rani, R.; Baltazar, F.; Kumar, V. Targeting monocarboxylate transporters (MCTs) in cancer: How close are we to the clinics? Semin. Cancer Biol. 2023, 90, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.A. An overview of patented small molecule stearoyl coenzyme-A desaturase inhibitors (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Tomita, N.; Kawakita, Y.; Ito, Y.; Ono, K.; Nii, N.; Miyazaki, T.; Yonemori, K.; Tawada, M.; Sumi, H.; et al. Discovery of novel and potent stearoyl Coenzyme A desaturase 1 (SCD1) inhibitors as anticancer agents. Bioorganic Med. Chem. 2017, 25, 3768–3779. [Google Scholar] [CrossRef]
- Meimaridou, E.; Kowalczyk, J.; Guasti, L.; Hughes, C.R.; Wagner, F.; Frommolt, P.; Nuernberg, P.; Mann, N.P.; Banerjee, R.; Saka, H.N.; et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat. Genet. 2012, 44, 740–742. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Reference | Carcinoma | Fundamental Observation |
|---|---|---|
| IN VITRO (cell culture) | ||
| [151] | lung carcinoma | MF-438 inhibition of SCD1 desaturase reduces cisplatin resistance in lung carcinoma 3D spheroids |
| [152] | melanoma | MF-438 inhibition of SCD1 desaturase reduces resistance to BRAFi plus MEKi inhibition of melanoma 3D spheroid formation |
| [167] | Glioblastoma(GBM | Merck inhibitors of SCD1 (Cpd3j) or FADS2 (SC26196) desaturases sensitize GBM cells to temozolomide-induced cell death in otherwise resistant cell lines |
| IN VIVO (tumor models) | ||
| [30] | melanoma | Etomoxir fatty acid oxidation inhibitor interferes with nongenetic xenograft resistance to BRAF inhibitors (in the presence of DCA perturbation of glycolytic regulation). |
| [119] | melanoma | Thioridazine fatty acid oxidation inhibitors robustly block nongenetic xenograft resistance to combinations of BRAF and MEK inhibitors. |
| [51] | pancreatic adenocarcinoma(PDAC) | Orlistat inhibition of fatty acid synthesis robustly blocks gemcitabine resistance in orthotopic xenografts. |
| [121] | pancreatic adenocarcinoma(PDAC) | Thioridazine substantially interferes with xenograft resistance to tumor-specific TCA cycle inhibitor, CPI-613. Crizotinib METi mimics this in vivo thioridazine effect, apparently through LMRS interference. |
| [156] | hepatocellular carcinoma (HCC) | Novel inhibitor (SSI-4) suppression of SCD1 activity robustly overcomes HCC xenograft resistance to multi-RTK inhibitor, sorafinib. |
| [164] | esophageal squamous cell carcinoma(ESCC) | MF-438 SCD1 inhibitor moderately enhances ESCC xenograft radiation sensitivity. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bingham, P.M.; Zachar, Z. Toward a Unifying Hypothesis for Redesigned Lipid Catabolism as a Clinical Target in Advanced, Treatment-Resistant Carcinomas. Int. J. Mol. Sci. 2023, 24, 14365. https://doi.org/10.3390/ijms241814365
Bingham PM, Zachar Z. Toward a Unifying Hypothesis for Redesigned Lipid Catabolism as a Clinical Target in Advanced, Treatment-Resistant Carcinomas. International Journal of Molecular Sciences. 2023; 24(18):14365. https://doi.org/10.3390/ijms241814365
Chicago/Turabian StyleBingham, Paul M., and Zuzana Zachar. 2023. "Toward a Unifying Hypothesis for Redesigned Lipid Catabolism as a Clinical Target in Advanced, Treatment-Resistant Carcinomas" International Journal of Molecular Sciences 24, no. 18: 14365. https://doi.org/10.3390/ijms241814365
APA StyleBingham, P. M., & Zachar, Z. (2023). Toward a Unifying Hypothesis for Redesigned Lipid Catabolism as a Clinical Target in Advanced, Treatment-Resistant Carcinomas. International Journal of Molecular Sciences, 24(18), 14365. https://doi.org/10.3390/ijms241814365