
The Regulatory Effect of Receptor-Interacting Protein Kinase 3 on CaMKIIδ in TAC-Induced Myocardial Hypertrophy
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
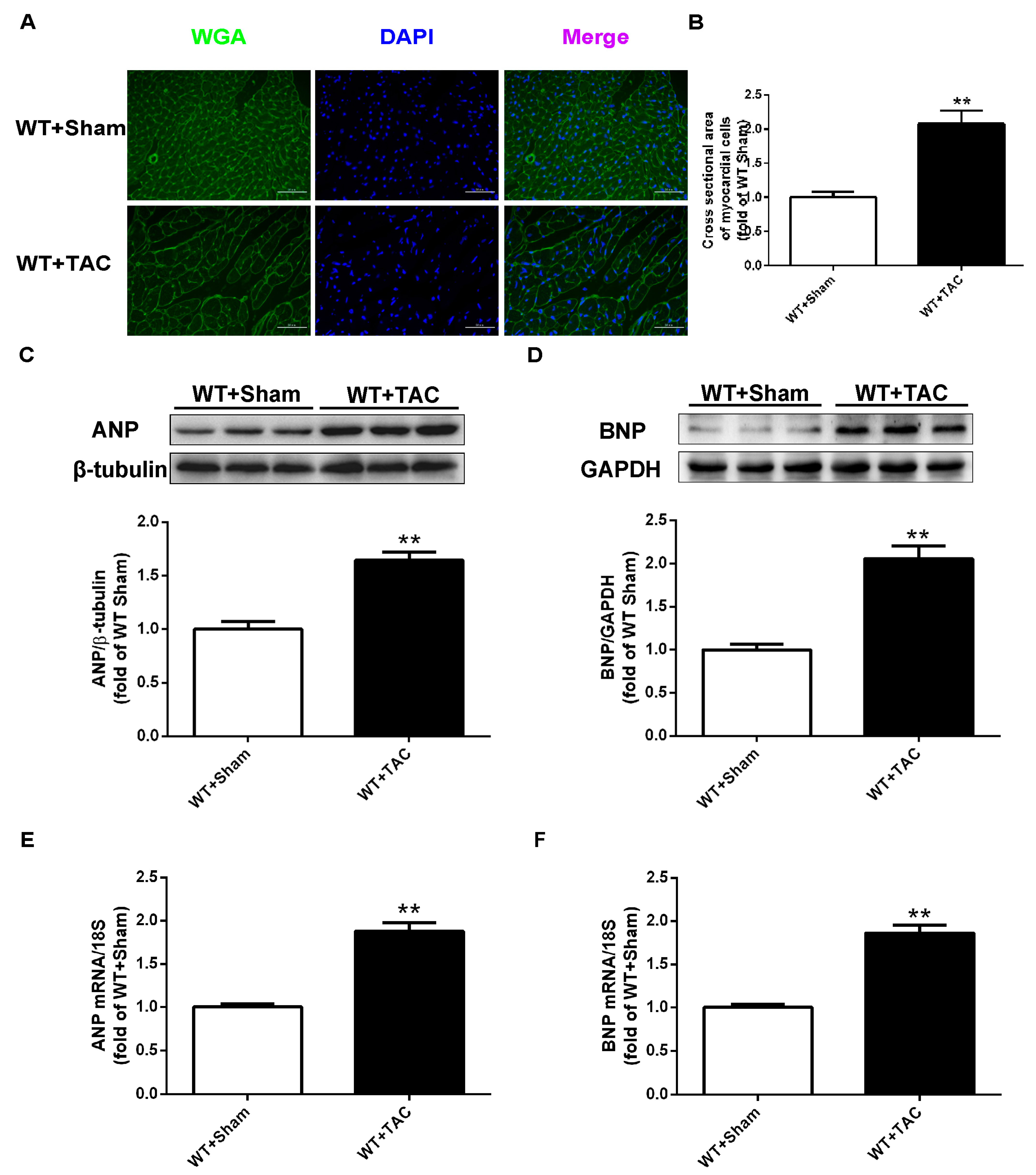
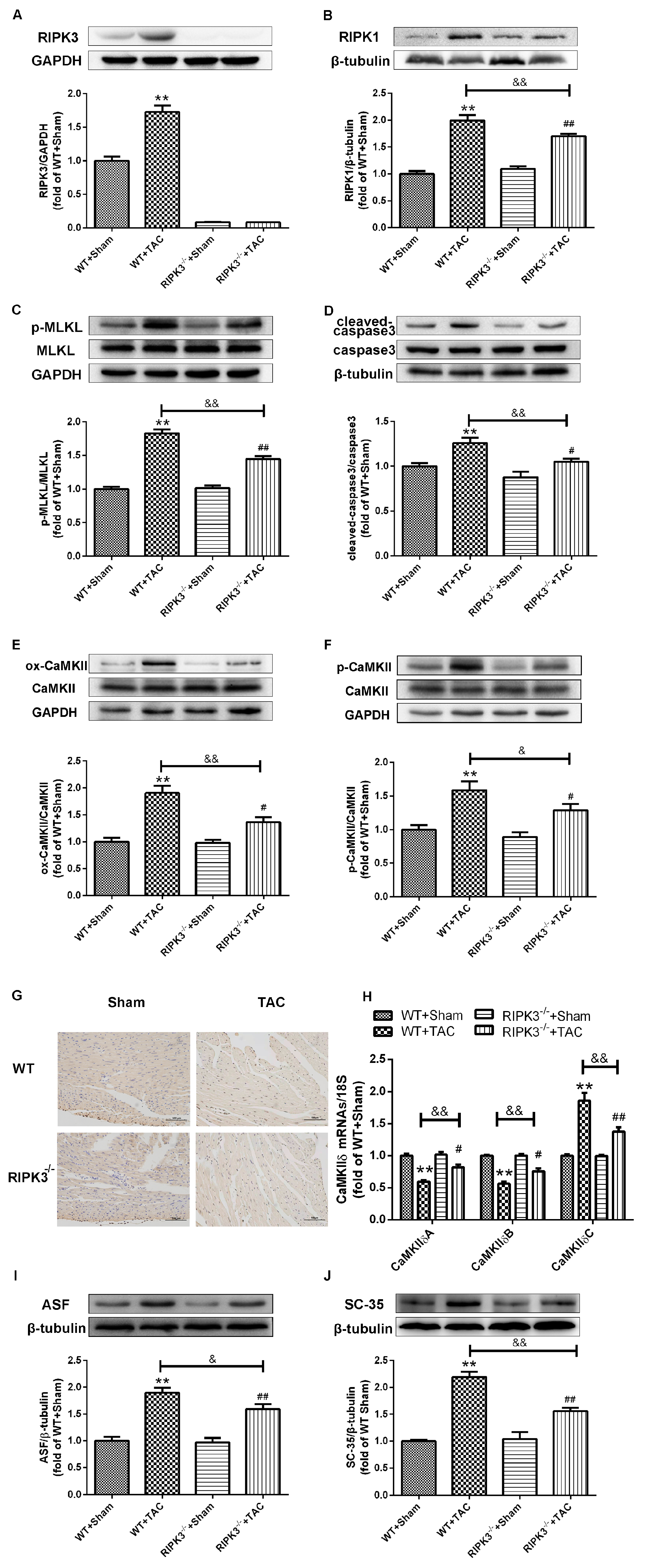
2.1. Cardiac Hypertrophy in Mice with Cardiac Dysfunction, Necroptosis, and CaMKIIδ Alternative Splicing Disorder
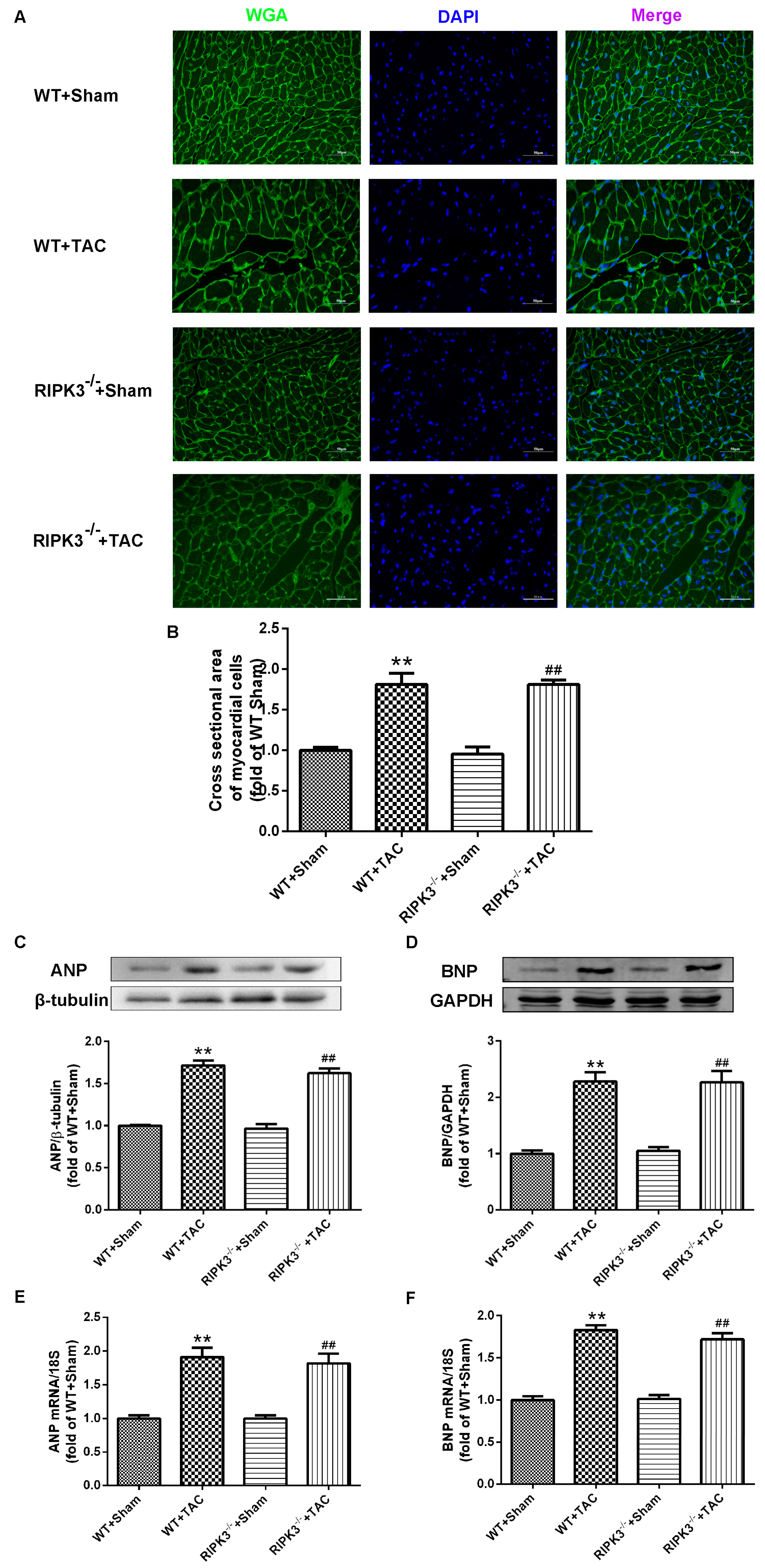
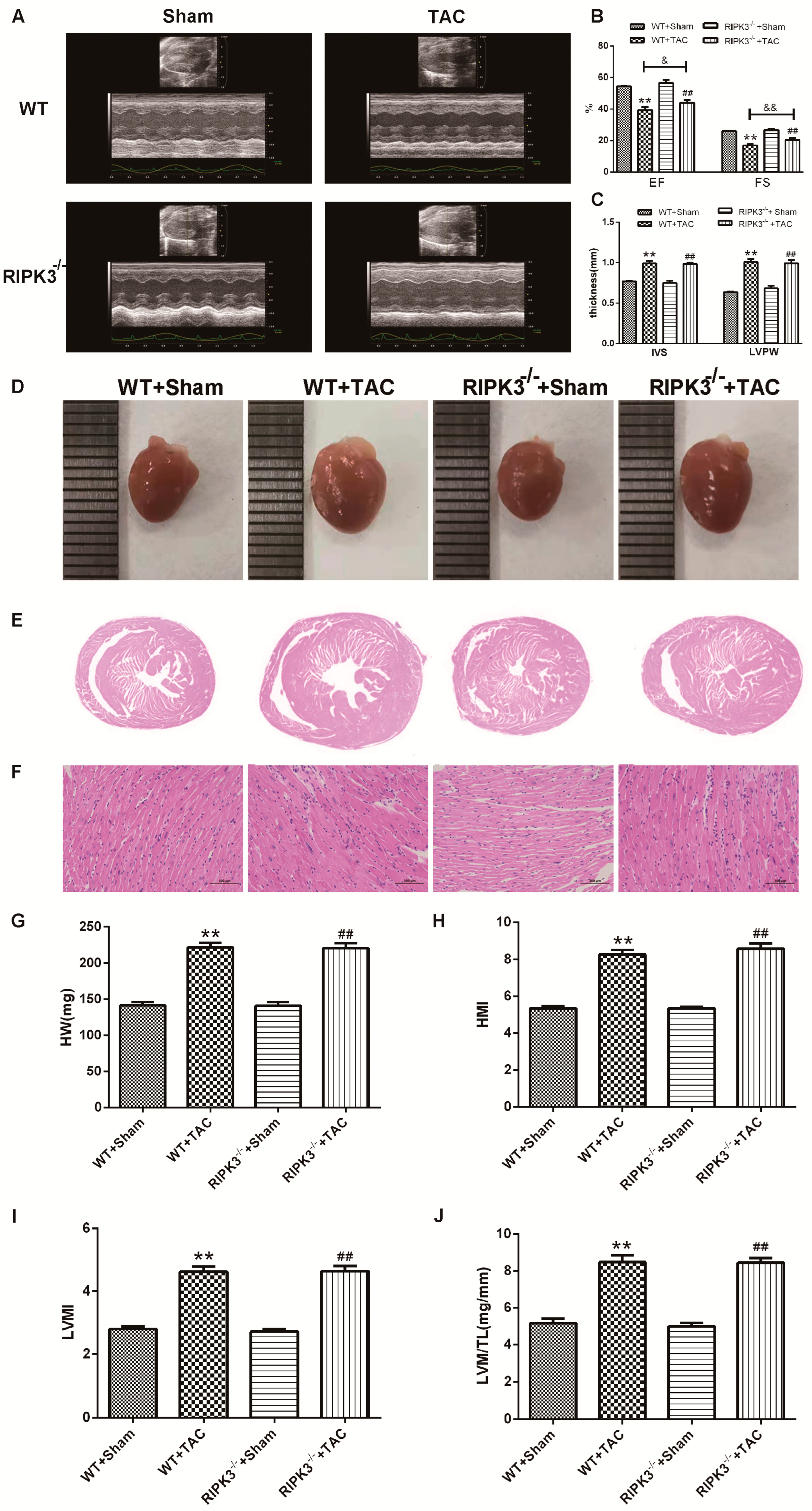
2.2. RIPK3 Deficiency Can Alleviate Cardiac Hypertrophy, Myocardial Injury, Myocardial Fibrosis, and Inflammation
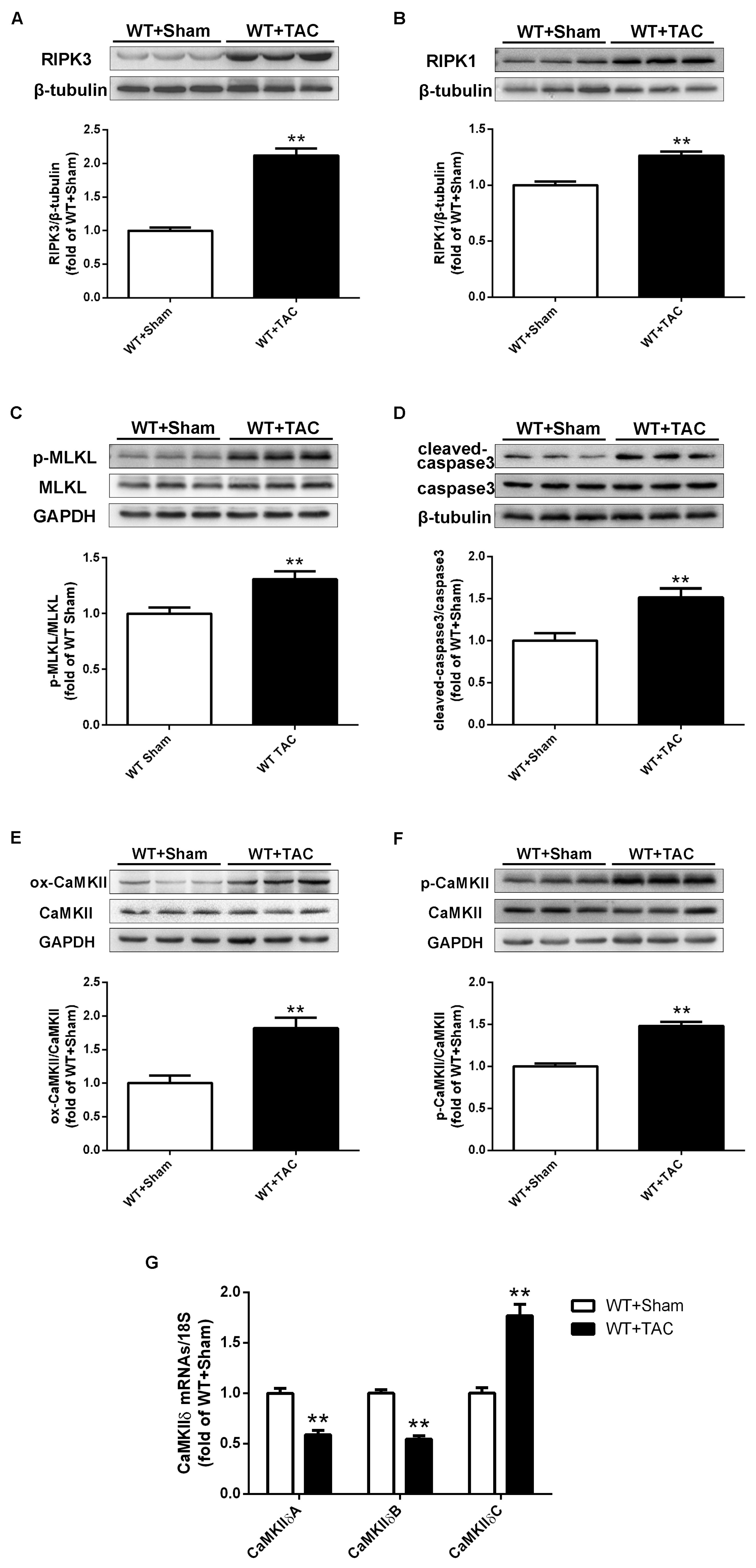
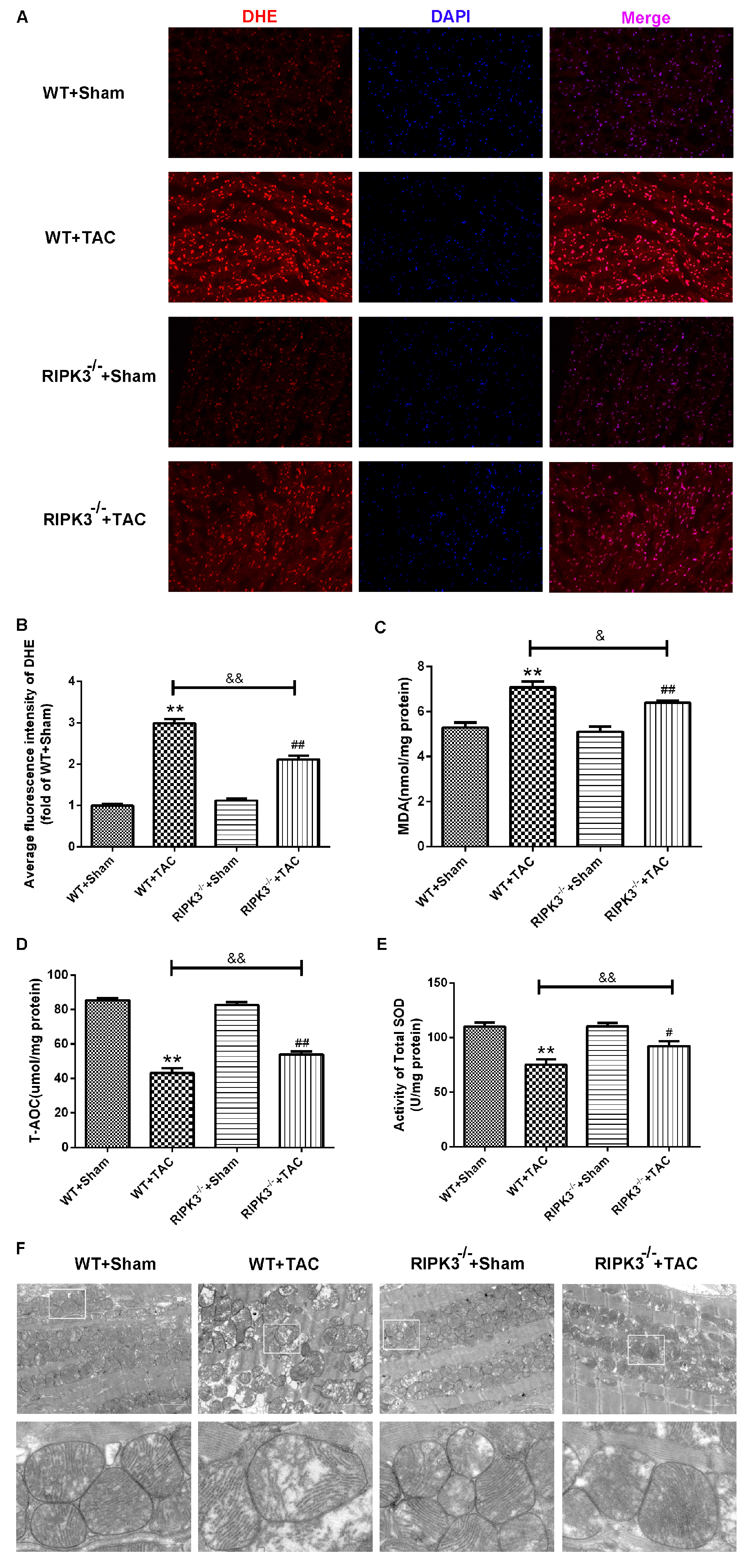
2.3. Loss of RIPK3 can Ameliorate Myocardial Necroptosis in Mice with Cardiac Hypertrophy, Regulate CaMKIIδ Splicing Disorder, Improve Oxidative Stress, and Ameliorate Myocardial Mitochondrial Ultrastructure
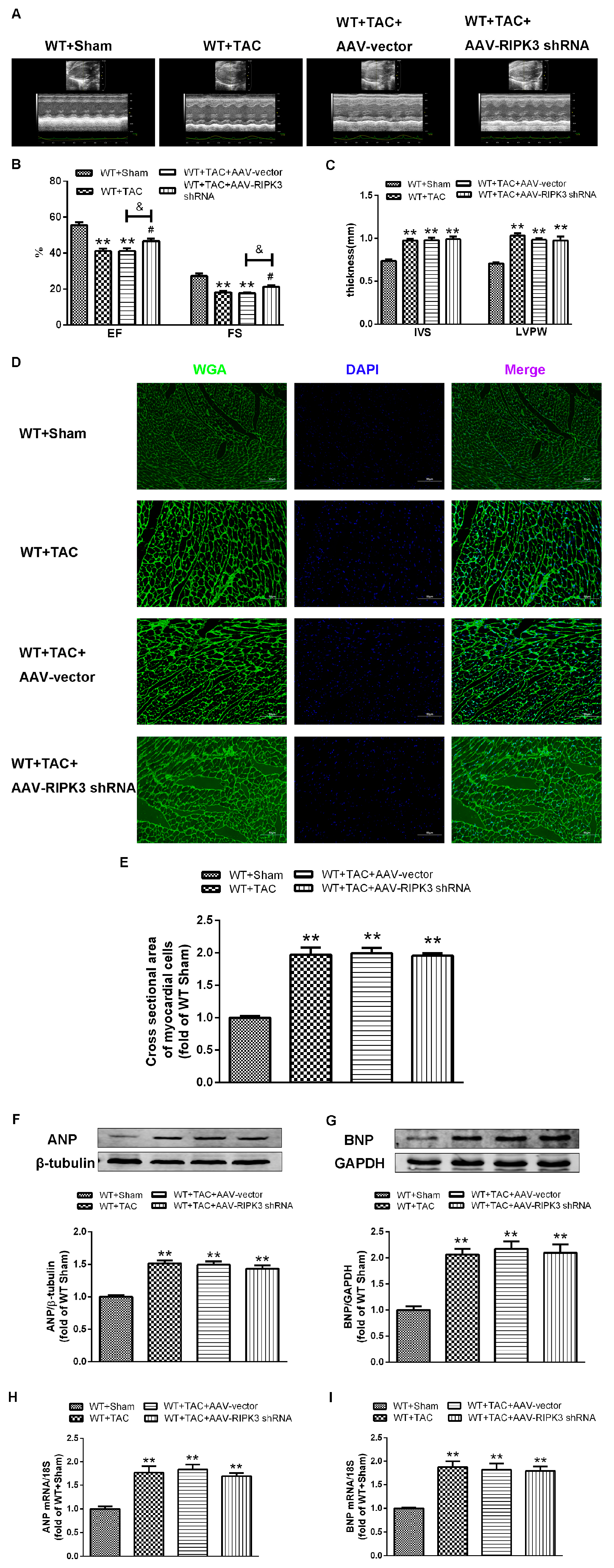
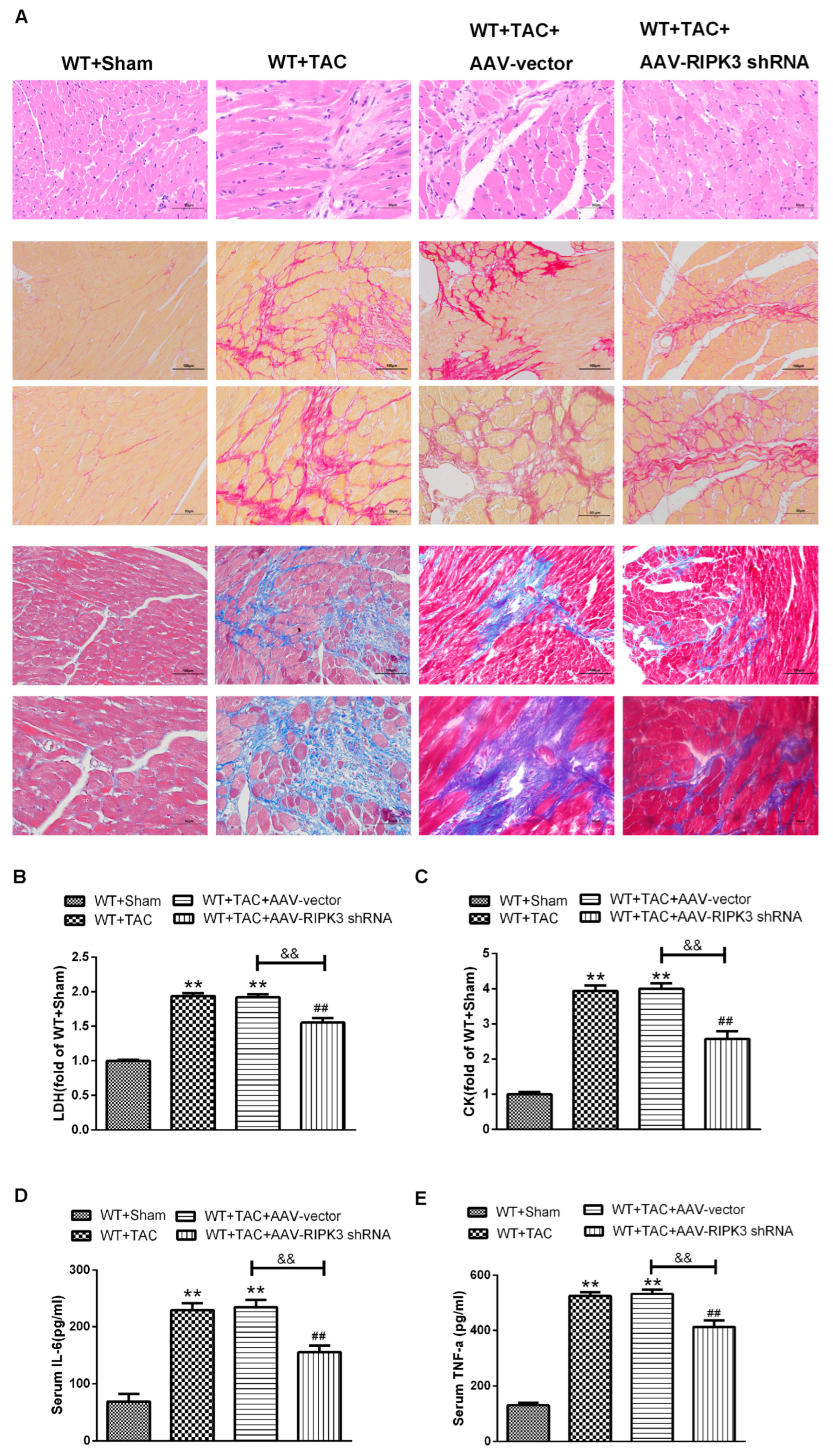
2.4. AAV-RIPK3 shRNA Interferes with the Expression of RIPK3, Reverses the Dysfunction of Myocardial Hypertrophy, Myocardial Fibrosis, and Inflammation, and Reduces Myocardial Damage
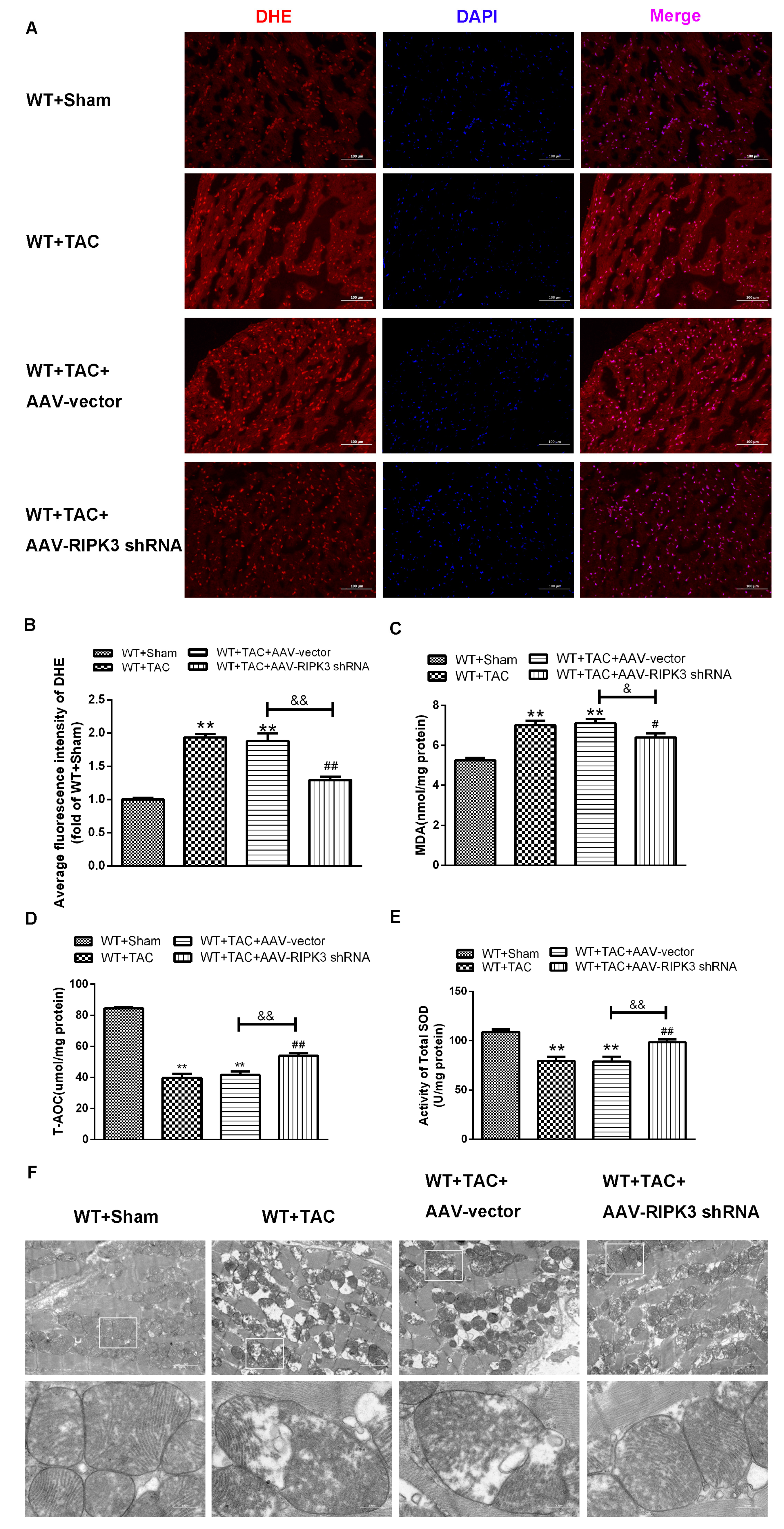
2.5. AAV-RIPK3 shRNA Interference Relieves Myocardial Necroptosis in Mice with Myocardial Hypertrophy, Regulates CaMKIIδ Splicing Disorder, Improves Oxidative Stress, and Ameliorates Myocardial Mitochondrial Ultrastructure
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Establishment of a Mouse Model of Myocardial Hypertrophy
4.3. Tail Vein Injection of AAV
4.4. Echocardiography
4.5. Heart Index Determination
4.6. Hematoxylin-Eosin (H&E) Staining
4.7. Sirius Scarlet Staining
4.8. Masson Staining
4.9. WGA Staining
4.10. TUNEL Staining
4.11. Measurement of Superoxide Formation
4.12. Determination of Blood Biochemical Indicators
4.13. Western Blotting Analysis
4.14. Quantitative Real-Time PCR (qRT-PCR)
4.15. Myocardial Ultrastructure Examination
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.-M.; Ma, Y.-T.; Yang, Y.-N.; Liu, F.; Chen, B.-D.; Han, W.; Zhang, J.-F.; Gao, X.-M. Downregulation of survival signalling pathways and increased apoptosis in the transition of pressure overload-induced cardiac hypertrophy to heart failure. Clin. Exp. Pharmacol. Physiol. 2009, 36, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Liu, J.; Liu, S.; Song, Q.; Liu, L.; Xie, L.; Han, Y.; Ji, Y. Hydrogen sulfide pretreatment improves mitochondrial function in myocardial hypertrophy via a SIRT3-dependent manner. Br. J. Pharmacol. 2018, 175, 1126–1145. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, I.; Hikoso, S.; Yamaguchi, O.; Kashiwase, K.; Nakai, A.; Takeda, T.; Watanabe, T.; Taniike, M.; Matsumura, Y.; Nishida, K.; et al. The antioxidant edaravone attenuates pressure overload-induced left ventricular hypertrophy. Hypertension 2005, 45, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yang, Y.; Xu, Z.; Lan, C.; Chen, C.; Li, C.; Chen, Z.; Yu, C.; Xia, X.; Liao, Q.; et al. Long Noncoding RNA Ahit Protects Against Cardiac Hypertrophy Through SUZ12 (Suppressor of Zeste 12 Protein Homolog)-Mediated Downregulation of MEF2A (Myocyte Enhancer Factor 2A). Circ. Heart Fail. 2020, 13, e006525. [Google Scholar] [CrossRef]
- Zhao, M.; Lu, L.; Lei, S.; Chai, H.; Wu, S.; Tang, X.; Bao, Q.; Chen, L.; Wu, W.; Liu, X. Inhibition of Receptor Interacting Protein Kinases Attenuates Cardiomyocyte Hypertrophy Induced by Palmitic Acid. Oxidative Med. Cell. Longev. 2016, 2016, 1451676. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Hua, Y.; Ding, Y.; Meng, G.; Zhang, W. RIPK3-Mediated Necroptosis in Diabetic Cardiomyopathy Requires CaMKII Activation. Oxidative Med. Cell. Longev. 2021, 2021, 6617816. [Google Scholar] [CrossRef]
- Chen, H.; Tang, L.-J.; Tu, H.; Zhou, Y.-J.; Li, N.-S.; Luo, X.-J.; Peng, J. Arctiin protects rat heart against ischemia/reperfusion injury via a mechanism involving reduction of necroptosis. Eur. J. Pharmacol. 2020, 875, 173053. [Google Scholar] [CrossRef]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Newton, K. RIPK1 and RIPK3: Critical regulators of inflammation and cell death. Trends Cell Biol. 2015, 25, 347–353. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Lawlor, K.E.; Murphy, J.M.; Vince, J.E. More to life than death: Molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr. Opin. Immunol. 2014, 26, 76–89. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wang, Z.; Ma, F.; Tran, B.; Zhong, R.; Xiong, Y.; Dai, T.; Wu, J.; Xin, X.; Guo, W.; et al. ZYZ-803 Mitigates Endoplasmic Reticulum Stress-Related Necroptosis after Acute Myocardial Infarction through Downregulating the RIP3-CaMKII Signaling Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 6173685. [Google Scholar] [CrossRef]
- Hegyi, B.; Bers, D.M.; Bossuyt, J. CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 127, 246–259. [Google Scholar] [CrossRef]
- Nickel, A.G.; Kohlhaas, M.; Bertero, E.; Wilhelm, D.; Wagner, M.; Sequeira, V.; Kreusser, M.M.; Dewenter, M.; Kappl, R.; Hoth, M.; et al. CaMKII does not control mitochondrial Ca(2+) uptake in cardiac myocytes. J. Physiol. 2020, 598, 1361–1376. [Google Scholar] [CrossRef]
- Chalmers, N.E.; Yonchek, J.; Steklac, K.E.; Ramsey, M.; Bayer, K.U.; Herson, P.S.; Quillinan, N. Calcium/Calmodulin-Dependent Kinase (CaMKII) Inhibition Protects Against Purkinje Cell Damage Following CA/CPR in Mice. Mol. Neurobiol. 2020, 57, 150–158. [Google Scholar] [CrossRef]
- Yang, Z.; Li, C.; Wang, Y.; Yang, J.; Yin, Y.; Liu, M.; Shi, Z.; Mu, N.; Yu, L.; Ma, H. Melatonin attenuates chronic pain related myocardial ischemic susceptibility through inhibiting RIP3-MLKL/CaMKII dependent necroptosis. J. Mol. Cell. Cardiol. 2018, 125, 185–194. [Google Scholar] [CrossRef]
- Feng, N.; Anderson, M.E. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J. Mol. Cell. Cardiol. 2017, 103, 102–109. [Google Scholar] [CrossRef]
- Sun, L.; Chen, Y.; Luo, H.; Xu, M.; Meng, G.; Zhang, W. Ca(2+)/calmodulin-dependent protein kinase II regulation by inhibitor 1 of protein phosphatase 1 alleviates necroptosis in high glucose-induced cardiomyocytes injury. Biochem. Pharmacol. 2019, 163, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.S. CaMKII delta overexpression in hypertrophy and heart failure: Cellular consequences for excitation-contraction coupling. Braz. J. Med. Biol. Res. = Rev. Bras. Pesqui. Medicas Biol. 2005, 38, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Luczak, E.D.; Anderson, M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell. Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Weinreuter, M.; Kreusser, M.M.; Beckendorf, J.; Schreiter, F.C.; Leuschner, F.; Lehmann, L.H.; Hofmann, K.P.; Rostosky, J.S.; Diemert, N.; Xu, C.; et al. CaM Kinase II mediates maladaptive post-infarct remodeling and pro-inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol. Med. 2014, 6, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Shioda, N.; Fukunaga, K. Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. Int. J. Mol. Sci. 2017, 19, 20. [Google Scholar] [CrossRef]
- Yao, J.; Qin, X.; Zhu, J.; Sheng, H. Dyrk1A-ASF-CaMKIIδ Signaling Is Involved in Valsartan Inhibition of Cardiac Hypertrophy in Renovascular Hypertensive Rats. Cardiology 2016, 133, 198–204. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, H.-Q.; Sun, L.-L.; Xu, M.-T.; Yu, J.; Liu, L.-L.; Zhang, J.-Y.; Wang, Y.-Q.; Wang, H.-X.; Bao, X.-F.; et al. Dihydromyricetin Attenuates Myocardial Hypertrophy Induced by Transverse Aortic Constriction via Oxidative Stress Inhibition and SIRT3 Pathway Enhancement. Int. J. Mol. Sci. 2018, 19, 2592. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Shao, M.; Zhuo, C.; Jiang, R.; Chen, G.; Shan, J.; Ping, J.; Tian, H.; Wang, L.; Lin, C.; Hu, L. Protective effect of hydrogen sulphide against myocardial hypertrophy in mice. Oncotarget 2017, 8, 22344–22352. [Google Scholar] [CrossRef]
- Schwarzer, M.; Osterholt, M.; Lunkenbein, A.; Schrepper, A.; Amorim, P.; Doenst, T. Mitochondrial reactive oxygen species production and respiratory complex activity in rats with pressure overload-induced heart failure. J. Physiol. 2014, 592, 3767–3782. [Google Scholar] [CrossRef]
- Cao, L.; Mu, W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol. Res. 2021, 163, 105297. [Google Scholar] [CrossRef]
- Linkermann, A.; Kunzendorf, U.; Krautwald, S. Phosphorylated MLKL causes plasma membrane rupture. Mol. Cell. Oncol. 2014, 1, e29915. [Google Scholar] [CrossRef]
- Wegner, K.W.; Saleh, D.; Degterev, A. Complex Pathologic Roles of RIPK1 and RIPK3: Moving Beyond Necroptosis. Trends Pharmacol. Sci. 2017, 38, 202–225. [Google Scholar] [CrossRef]
- Marunouchi, T.; Nishiumi, C.; Iinuma, S.; Yano, E.; Tanonaka, K. Effects of Hsp90 inhibitor on the RIP1-RIP3-MLKL pathway during the development of heart failure in mice. Eur. J. Pharmacol. 2021, 898, 173987. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef]
- Hu, Y.; Pan, H.; Peng, J.; He, J.; Tang, M.; Yan, S.; Rong, J.; Li, J.; Zheng, Z.; Wang, H.; et al. Resveratrol inhibits necroptosis by mediating the TNF-α/RIP1/RIP3/MLKL pathway in myocardial hypoxia/reoxygenation injury. Acta Biochim. Biophys. Sin. 2021, 53, 430–437. [Google Scholar] [CrossRef]
- Mohammed, S.; Nicklas, E.H.; Thadathil, N.; Selvarani, R.; Royce, G.H.; Kinter, M.; Richardson, A.; Deepa, S.S. Role of necroptosis in chronic hepatic inflammation and fibrosis in a mouse model of increased oxidative stress. Free Radic. Biol. Med. 2021, 164, 315–328. [Google Scholar] [CrossRef]
- Jiang, S.J.; Wang, W. Research progress on the role of CaMKII in heart disease. Am. J. Transl. Res. 2020, 12, 7625–7639. [Google Scholar] [PubMed]
- Rusciano, M.R.; Sommariva, E.; Douin-Echinard, V.; Ciccarelli, M.; Poggio, P.; Maione, A.S. CaMKII Activity in the Inflammatory Response of Cardiac Diseases. Int. J. Mol. Sci. 2019, 20, 4374. [Google Scholar] [CrossRef] [PubMed]
- Baier, M.J.; Klatt, S.; Hammer, K.P.; Maier, L.S.; Rokita, A.G. Ca(2+)/calmodulin-dependent protein kinase II is essential in hyperacute pressure overload. J. Mol. Cell. Cardiol. 2020, 138, 212–221. [Google Scholar] [CrossRef]
- Sossalla, S.; Fluschnik, N.; Schotola, H.; Ort, K.R.; Neef, S.; Schulte, T.; Wittköpper, K.; Renner, A.; Schmitto, J.D.; Gummert, J.; et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ. Res. 2010, 107, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Purohit, A.; Rokita, A.G.; Guan, X.; Chen, B.; Koval, O.M.; Voigt, N.; Neef, S.; Sowa, T.; Gao, Z.; Luczak, E.D.; et al. Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 2013, 128, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Dybkova, N.; Sedej, S.; Napolitano, C.; Neef, S.; Rokita, A.G.; Hünlich, M.; Brown, J.H.; Kockskämper, J.; Priori, S.G.; Pieske, B.; et al. Overexpression of CaMKIIδc in RyR2R4496C+/− knock-in mice leads to altered intracellular Ca2+ handling and increased mortality. J. Am. Coll. Cardiol. 2011, 57, 469–479. [Google Scholar] [CrossRef]
- Duran, J.; Nickel, L.; Estrada, M.; Backs, J.; van den Hoogenhof, M.M.G. CaMKIIδ Splice Variants in the Healthy and Diseased Heart. Front. Cell Dev. Biol. 2021, 9, 644630. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Cao, W.; Liao, R.; Wang, J.; Wang, Y.; Tong, L.; Chen, X.; Zhu, W.; Zhang, W. PP1γ functionally augments the alternative splicing of CaMKIIδ through interaction with ASF. Am. J. Physiol. Cell Physiol. 2014, 306, C167–C177. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Ding, J.H.; Byeon, C.W.; Kim, J.H.; Hertel, K.J.; Jeong, S.; Fu, X.D. SR proteins induce alternative exon skipping through their activities on the flanking constitutive exons. Mol. Cell. Biol. 2011, 31, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Shi, H.; Zhang, F.; Li, H.; Han, Q. RIP3 Contributes to Cardiac Hypertrophy by Influencing MLKL-Mediated Calcium Influx. Oxid. Med. Cell. Longev. 2022, 2022, 5490553. [Google Scholar] [CrossRef]
- Xiang, D.; Liu, Y.; Zhou, S.; Zhou, E.; Wang, Y. Protective Effects of Estrogen on Cardiovascular Disease Mediated by Oxidative Stress. Oxid. Med. Cell. Longev. 2021, 2021, 5523516. [Google Scholar] [CrossRef]
- Song, S.; Ding, Y.; Dai, G.-L.; Zhang, Y.; Xu, M.-T.; Shen, J.-R.; Chen, T.-T.; Chen, Y.; Meng, G.-L. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 2021, 42, 230–241. [Google Scholar] [CrossRef]
- Song, X.; Li, T. Ripk3 mediates cardiomyocyte necrosis through targeting mitochondria and the JNK-Bnip3 pathway under hypoxia-reoxygenation injury. J. Recept. Signal Transduct. Res. 2019, 39, 331–340. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, J.; Zhang, J.; Cao, J.; Wang, X.; Zhang, W.; Chen, X. The Regulatory Effect of Receptor-Interacting Protein Kinase 3 on CaMKIIδ in TAC-Induced Myocardial Hypertrophy. Int. J. Mol. Sci. 2023, 24, 14529. https://doi.org/10.3390/ijms241914529
Qian J, Zhang J, Cao J, Wang X, Zhang W, Chen X. The Regulatory Effect of Receptor-Interacting Protein Kinase 3 on CaMKIIδ in TAC-Induced Myocardial Hypertrophy. International Journal of Molecular Sciences. 2023; 24(19):14529. https://doi.org/10.3390/ijms241914529
Chicago/Turabian StyleQian, Jianan, Jingjing Zhang, Ji Cao, Xue Wang, Wei Zhang, and Xiangfan Chen. 2023. "The Regulatory Effect of Receptor-Interacting Protein Kinase 3 on CaMKIIδ in TAC-Induced Myocardial Hypertrophy" International Journal of Molecular Sciences 24, no. 19: 14529. https://doi.org/10.3390/ijms241914529