
Integrated Transcriptomic and Metabolomics Analyses Reveal Molecular Responses to Cold Stress in Coconut (Cocos nucifera L.) Seedlings

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
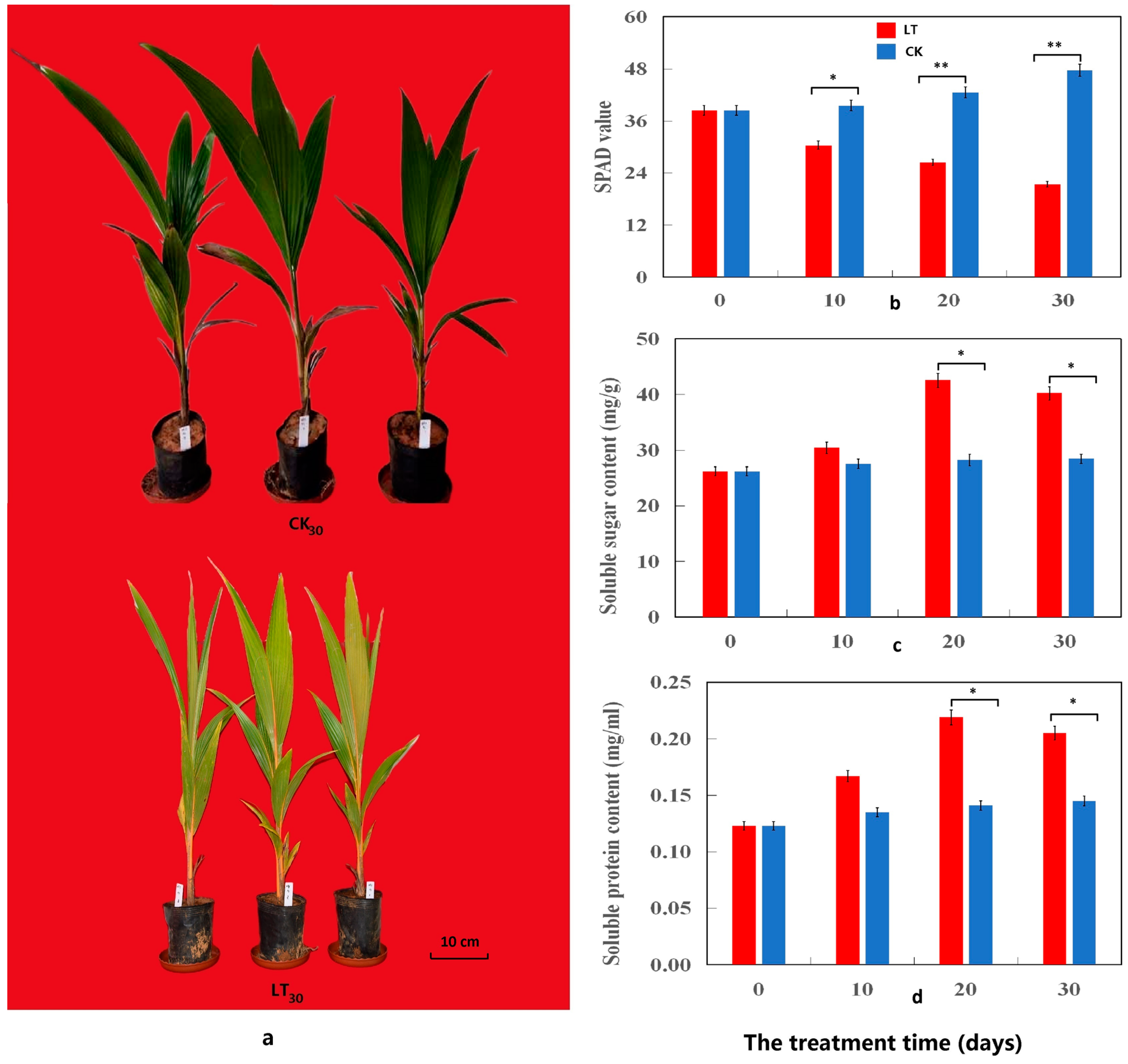
2.1. Physiological Differences in Response to Cold Stress
2.2. Transcriptome Analysis
2.2.1. Evaluation of Transcriptome Sequencing Data
2.2.2. The Major Regulator Genes of Significantly Enriched KEGG Pathways in Response to Cold Stress in Coconut
2.2.3. The Core Genes and Transcription Factors in Response to Cold Stress in Coconut
2.2.4. qRT-PCR Validation
2.3. Metabolome Analysis
2.3.1. Metabolite Profiles of Coconut in Response to Cold Stress
2.3.2. Differentially Accumulated Metabolites (DAMs) in Response to Low Temperatures
2.4. Correlation Analysis between DEGs and DAMs
2.4.1. Amino Acid Metabolism Considerably Contributes to Cold Stress in Coconut
2.4.2. Carbohydrate, Lipid, and Nucleotide Metabolism Contributes to Cold Stress in Coconut
2.4.3. Biosynthesis of Other Secondary Metabolites and Metabolism of Cofactors and Vitamins Contributes to Cold Tolerance in Coconut
2.4.4. Translation and Membrane Transport Contributes to Cold Stress in Coconut
3. Discussion
3.1. Physiological Response of Coconut under Cold Stress
3.2. Transcription Factor in Response to Cold Stress in Coconut
3.3. The Core Regulator Genes in Response to Cold Stress in Coconut
3.4. The Major Significant Enriched KEGG Pathways in Transcription and Metabolism in Response to Cold Stress in Coconut
3.5. Combined Analysis of Transcription and Metabolism in Response to Cold Stress in Coconut
3.5.1. Amino Acid Metabolism in Response to Cold Stress in Coconut
3.5.2. Carbohydrate Metabolism in Response to Cold Stress in Coconut
3.5.3. Flavonoid Metabolism in Response to Cold Stress in Coconut
3.5.4. Lipid Metabolism in Response to Cold Stress in Coconut
3.5.5. Nucleotide Metabolism in Response to Cold Stress in Coconut
4. Materials and Methods
4.1. Plant Materials and Cold Treatments
4.2. Measurements of Plant Height, Dry Weight, and Soil and SPAD Values
4.3. Physiological Index Measurements
4.4. RNA Extraction and RNA-Sequencing (RNA-Seq)
4.5. Metabolite Analysis
4.6. Integrated Metabolome and Transcriptome Analyses
4.7. Quantitative Real-Time PCR (qRT-PCR) Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwon, K.; Park, K.H.; Rhee, K.C. Fractionation and characterization of proteins from coconut (Cocos nucifera L.). J. Agric. Food Chem. 1996, 44, 1741–1745. [Google Scholar] [CrossRef]
- Peamprasart, T.; Chiewchan, N. Effect of fat content and preheat treatment on the apparent viscosity of coconut milk after homogenization. J. Food Eng. 2006, 77, 653–658. [Google Scholar] [CrossRef]
- Debmandal, M.; Mandal, S. Coconut (Cocos nucifera L.: Arecaceae): In health promotion and disease prevention. Asian Pac. J. Trop. Med. 2011, 4, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.L.; Li, J.; Wang, B.Z. Overview of comprehensive research progress of coconut in China. Trop. Agric. China 2007, 5, 30–31. (In Chinese) [Google Scholar]
- Hebbar, K.B.; Balasimha, D.; Thomas, G.V. Plantation crops response to climate change: Coconut perspective. In Climate-Resilient Horticulture: Adaptation and Mitigation Strategies; Singh, H., Rao, N., Eds.; Springer: New Delhi, India, 2013; pp. 177–187. [Google Scholar]
- Zhao, S.L.; Cao, H.X. Collection, Conservation, Identification, Evaluation and Innovative Utilization of Coconut Germplasm Resources, 1st ed.; China Agriculture Press: Beijing, China, 2012; pp. 112–116. [Google Scholar]
- Cao, H.; Huang, H.; Lei, X.; Zhang, D.; Zhang, R. Difference of the leaf anatomical structure of coconut varieties under low temperature treatments. Chin. J. Trop. Crops 2014, 35, 2420–2425. (In Chinese) [Google Scholar]
- Yang, Y.; Iqbal, A.; Qadri, R. Breeding of coconut (Cocos nucifera L.): The tree of life. In Advances in Plant Breeding Strategies: Fruits; Al-Khayri, J., Jain, S., Johnson, D., Eds.; Springer: Cham, Switzerland, 2018; pp. 673–725. [Google Scholar]
- Zhao, Y.; Zhou, M.; Xu, K.; Li, J.H.; Li, S.S.; Zhang, S.H.; Yang, X.J. Integrated transcriptomics and metabolomics analyses provide insights into cold stress response in wheat. Crop J. 2019, 7, 857–866. [Google Scholar] [CrossRef]
- Jin, J.J.; Zhang, H.; Zhang, J.F.; Liu, P.P.; Chen, X.; Li, Z.F.; Xu, Y.L.; Lu, P.; Cao, P.J. Integrated transcriptomics and metabolomics analysis to characterize cold stress responses in Nicotiana tabacum. BMC Genom. 2017, 18, 496. [Google Scholar] [CrossRef]
- Hu, Y.M.; Peng, X.J.; Wang, F.F.; Chen, P.L.; Zhao, M.L.; Shen, S.H. Natural population re-sequencing detects the genetic basis of local adaptation to low temperature in a woody plant. Plant Mol. Biol. 2021, 105, 585–599. [Google Scholar] [CrossRef]
- Theocharis, A.; Cl’ement, C.; Barka, E.A. Physiological and molecular changes in plants grown at low temperatures. Planta 2012, 235, 1091–1105. [Google Scholar] [CrossRef]
- Bailey-Serres, J.; Mittler, R. The roles of reactive oxygen species in plant cells. Plant Physiol. 2006, 141, 311. [Google Scholar] [CrossRef]
- Meng, D.; Yu, X.; Ma, L.; Hu, J.; Liang, Y.; Liu, X.; Yin, H.; Liu, H.; He, X.; Li, D. Transcriptomic Response of Chinese Yew (Taxus chinensis) to Cold Stress. Front. Plant Sci. 2017, 8, 468. [Google Scholar] [CrossRef]
- Sun, S.; Fang, J.; Lin, M.; Hu, C.; Qi, X.; Chen, J.; Zhong, Y.; Muhammad, A.; Li, Z.; Li, Y. Comparative Metabolomic and Transcriptomic Studies Reveal Key Metabolism Pathways Contributing to Freezing Tolerance Under Cold Stress in Kiwifruit. Front. Plant Sci. 2021, 12, 628969. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, V.; Shahzad, B.; Ramakrishnan, M.; Singh Sidhu, G.P.; Bali, A.S.; Handa, N.; Kapoor, D.; Yadav, P.; Khanna, K.; et al. Photosynthetic response of plants under different abiotic stresses: A review. J. Plant Growth Regul. 2020, 39, 509–531. [Google Scholar] [CrossRef]
- Sinha, S.; Kukreja, B.; Arora, P.; Sharma, M.; Pandey, G.K.; Agarwal, M.; Chinnusamy, V. The omics of cold stress responses in plants. In Elucidation of Abiotic Stress Signaling in Plants: Functional Genomics Perspectives; Pandey, G.K., Ed.; Springer: New York, NY, USA, 2015; pp. 143–194. [Google Scholar] [CrossRef]
- Miura, K.; Furumoto, T. Cold signaling and cold response in plants. Int. J. Mol. Sci. 2013, 14, 5312–5337. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Fakher, B.; Ashraf, M.A.; Cheng, Y.; Wang, B.; Qin, Y. Plant low-temperature stress: Signaling and response. Agronomy 2022, 12, 702. [Google Scholar] [CrossRef]
- Ma, L.; Coulter, J.A.; Liu, L.J.; Zhao, Y.H.; Chang, Y.; Pu, Y.Y.; Zeng, X.C.; Xu, Y.Z.; Wu, J.Y.; Fang, Y.; et al. Transcriptome analysis reveals key cold-stress-responsive genes in winter rapeseed (Brassica rapa L.). Int. J. Mol. Sci. 2019, 20, 1071. [Google Scholar] [CrossRef] [PubMed]
- Fürtauer, L.; Weiszmann, J.; Weckwerth, W.; Nägele, T. Dynamics of plant metabolism during cold acclimation. Int. J. Mol. Sci. 2019, 20, 5411. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.A.; Mahajan, R.; Pakhtoon, M.M.; Urwat, U.; Bashir, Z.; Shah, A.A.; Agrawal, A.; Bhat, B.; Sofi, P.A.; Masi, A.; et al. Low temperature stress tolerance: An insight into the omics approaches for legume crops. Front. Plant Sci. 2022, 13, 888710. [Google Scholar] [CrossRef] [PubMed]
- Halman, J.M.; Schaberg, P.G.; Hawley, G.J.; Eagar, C. Calcium addition at the Hubbard Brook Experimental Forest increases sugar storage, antioxidant activity and cold tolerance in native red spruce (Picea rubens). Tree Physiol. 2008, 28, 855–862. [Google Scholar] [CrossRef]
- George, I.S.; Pascovici, D.; Mirzaei, M.; Haynes, P.A. Quantitative proteomic analysis of cabernet sauvignon grape cells exposed to thermal stresses reveals alterations in sugar and phenylpropanoid metabolism. Proteomics 2015, 15, 3048–3060. [Google Scholar] [CrossRef]
- Zhu, A.D.; Li, W.Y.; Ye, J.L.; Sun, X.H.; Ding, Y.D.; Cheng, Y.J.; Deng, X.X. Microarray expression profiling of postharvest ponkan mandarin (Citrus reticulata) fruit under cold storage reveals regulatory gene candidates and implications on soluble sugars metabolism. J. Int. Plant Biol. 2011, 53, 358–374. [Google Scholar] [CrossRef]
- Taïbi, K.; Del Campo, A.D.; Vilagrosa, A.; Bellés, J.M.; López-Gresa, M.; López-Nicolás, J.M.; Mulet, J.M. Distinctive physiological and molecular responses to cold stress among cold-tolerant and cold-sensitive Pinus halepensis seed sources. BMC Plant Biol. 2018, 18, 236. [Google Scholar] [CrossRef] [PubMed]
- Macháècková, I.; Hanièová, A.; Krekule, J. Levels of ethylene, ACC, MACC, ABA and proline as indicators of cold hardening and frost resistance in winter wheat. Physiol. Plant. 2010, 76, 603–607. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signalling. Biol. Plant. 2020, 54, 201–212. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, N.; Jia, Z.K.; Duan, J.; Chen, F.J.; Sang, Z.Y.; Ma, L.Y. Effect of exogenous abscisic acid on cold acclimation in two Magnolia species. Biol. Plant. 2016, 60, 555–562. [Google Scholar] [CrossRef]
- Kim, T.E.; Kim, S.K.; Han, T.J.; Lee, J.S.; Chang, S.C. ABA and polyamines act independently in primary leaves of cold-stressed tomato (Lycopersicon esculentum). Physiol. Plant. 2010, 115, 370–376. [Google Scholar] [CrossRef]
- Du, H.; Liu, H.B.; Xiong, L.Z. Endogenous auxin and jasmonic acid levels are differentially modulated by abiotic stresses in rice. Front. Plant Sci. 2013, 4, 397. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, J.P.; Zhang, Y.X.; Yan, X.R. Integrated transcriptomic and metabolomic analyses of yellow horn (Xanthoceras sorbifolia) in response to cold stress. PLoS ONE 2020, 15, e0236588. [Google Scholar] [CrossRef]
- Suzuki, N. Temperature stress and responses in plants. Int. J. Mol. Sci. 2019, 20, 2001. [Google Scholar] [CrossRef]
- Yuan, P.; Yang, T.; Poovaiah, B.W. Calcium signaling-mediated plant response to cold stress. Int. J. Mol. Sci. 2018, 19, 3896. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Ohta, M.; Kanrar, S.; Lee, B.H.; Hong, X.H.; Agarwal, M.; Zhu, J.K. ICE1: A regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev. 2003, 17, 1043–1054. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, M.; Lee, J.H.; Lee, H.J.; Park, C.M. The unified ICE-CBF pathway provides a transcriptional feedback control of freezing tolerance during cold acclimation in Arabidopsis. Plant Mol. Biol. 2015, 89, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Zhao, L.; Ren, Y.; Yang, S.; Zhu, J.K.; Zhao, C. The transcription factor ICE1 functions in cold stress response by binding to the promoters of CBF and COR genes. J. Integr. Plant Biol. 2020, 62, 258–263. [Google Scholar] [CrossRef]
- Zuther, E.; Buchel, K.; Hundertmark, M.; Stitt, M.; Hincha, D.K.; Heyer, A.G. The role of raffinose in the cold acclimation response of Arabidopsis thaliana. FEBS Lett. 2004, 576, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, Y.; Lu, J.; Shao, H. Roles of plant soluble sugars and their responses to plant cold stress. Afr. J. Biotechnol. 2009, 8, 2004–2010. [Google Scholar]
- Mehmood, S.S.; Lu, G.; Luo, D.; Hussain, M.A.; Raza, A.; Zafar, Z.; Zhang, X.K.; Cheng, Y.; Zou, X.L.; Lv, Y. Integrated analysis of transcriptomics and proteomics provides insights into the molecular regulation of cold response in Brassica napus. Environ. Exp. Bot. 2021, 187, 104480. [Google Scholar] [CrossRef]
- Sinha, S.; Raxwal, V.K.; Joshi, B.; Jagannath, A.; Katiyar-Agarwal, S.; Goel, S.; Kumar, A.; Agarwal, M. De novo transcriptome profiling of cold-stressed siliques during pod filling stages in Indian mustard (Brassica juncea L.). Front. Plant Sci. 2015, 6, 932. [Google Scholar] [CrossRef]
- Zhang, W.J.; Xu, H.Y.; Duan, X.X.; Hu, J.; Li, J.J.; Zhao, L.; Ma, Y.P. Characterizing the leaf transcriptome of Chrysanthemum rhombifolium (Ling et C. Shih), a drought resistant, endemic plant from China. Front. Genet. 2021, 12, 625985. [Google Scholar] [CrossRef] [PubMed]
- Klepikova, A.V.; Kulakovskiy, I.V.; Kasianov, A.S.; Logacheva, M.D.; Penin, A.A. An update to database TraVA: Organ-specific cold stress response in Arabidopsis thaliana. BMC Plant Biol. 2019, 19, 49. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.; Han, Z.K.; Chen, Y.N.; Huai, D.X.; Kang, Y.P.; Wang, Z.H.; Yan, L.Y.; Jiang, H.F.; Lei, Y.; et al. Integrated transcriptomics and metabolomics analysis reveal key metabolism pathways contributing to cold tolerance in peanut. Front. Plant Sci. 2021, 12, 752474. [Google Scholar] [CrossRef]
- Xu, H.; Li, J.; Wang, L.; Li, X.; Liu, Y.; Wang, X.; Gao, T.; Ma, Y. Integrated transcriptomic and metabolomics analysis reveals abscisic acid signal transduction and sugar metabolism pathways as defense responses to cold stress in Argyranthemum frutescens. Environ. Exp. Bot. 2023, 205, 105115. [Google Scholar] [CrossRef]
- Licausi, F.; Ohme-Takagi, M.; Perata, P. APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: Mediators of stress responses and developmental programs. New Phytol. 2013, 199, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Shiryaeva, L.; Antti, H.; Schroder, W.P.; Strimbeck, R.; Shiriaev, A.S. Pair-wise multicomparison and OPLS analyses of cold-acclimation phases in Siberian spruce. Metabolomics 2012, 8, 123–130. [Google Scholar] [CrossRef]
- Perotti, V.E.; Moreno, A.S.; Trípodi, K.E.; Meier, G.; Bello, F.; Cocco, M. Proteomic and metabolomic profiling of Valencia orange fruit after natural frost exposure. Physiol. Plant. 2015, 153, 337–354. [Google Scholar] [CrossRef]
- Pagter, M.; Alpers, J.; Erban, A.; Kopka, J.; Zuther, E.; Hincha, D.K. Rapid transcriptional and metabolic regulation of the deacclimation process in cold acclimated Arabidopsis thaliana. BMC Genom. 2017, 18, 731. [Google Scholar] [CrossRef]
- Jiang, J.; Hou, R.; Yang, N.; Li, L.; Deng, J.; Qin, G.; Ding, D. Physiological and TMT-labeled proteomic analyses reveal important roles of sugar and secondary metabolism in Citrus junos under cold stress. J. Proteom. 2021, 237, 104145. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, H.; Ren, J.; Dong, J.; Zhao, X.; Wang, X.; Wang, J.; Zhong, C.; Zhao, S.; Liu, X.; et al. Comparative transcriptome-based mining and expression profiling of transcription factors related to cold tolerance in peanut. Int. J. Mol. Sci. 2020, 21, 1921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, C.; Ren, J.; Dong, J.; Shi, X.; Zhao, X.; Wang, X.; Wang, J.; Zhong, C.; Zhao, S.; et al. Anadvanced lipid metabolism system revealed by transcriptomic and lipidomic analyses plays a central role in peanut cold tolerance. Front. Plant Sci. 2020, 11, 1110. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Su, W.; Hussain, M.A.; Mehmood, S.S.; Zhang, X.; Cheng, Y.; Zou, X.; Lv, Y. Integrated Analysis of Metabolome and Transcriptome Reveals Insights for Cold Tolerance in Rapeseed (Brassica napus L.). Front. Plant Sci. 2021, 12, 721681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, L.; Xie, Y.; Zhao, Z.; Su, L.; Tang, Y.; Sun, B.; Lai, Y.; Li, H. Transcriptome and Metabolome Analyses Reveal Molecular Responses of Two Pepper (Capsicum annuum L.) Cultivars to Cold Stress. Front. Plant Sci. 2022, 13, 819630. [Google Scholar] [CrossRef]
- Gao, C.; Mumtaz, M.A.; Zhou, Y.; Yang, Z.; Shu, H.; Zhu, J.; Bao, W.; Cheng, S.; Yin, L.; Huang, J.; et al. Integrated Transcriptomic and Metabolomic Analyses of Cold-Tolerant and Cold-Sensitive Pepper Species Reveal Key Genes and Essential Metabolic Pathways Involved in Response to Cold Stress. Int. J. Mol. Sci. 2022, 23, 6683. [Google Scholar] [CrossRef]
- Li, F.; Lu, X.; Duan, P.; Liang, Y.; Cui, J. Integrating transcriptome and metabolome analyses of the response to cold stress in pumpkin (Cucurbita maxima). PLoS ONE 2021, 16, e0249108. [Google Scholar] [CrossRef]
- Hu, Z.R.; Yan, W.J.; Yang, C.K.; Huang, X.B.; Hu, X.T.; Li, Y.Y.; Yang, J.S.; Xiang, S.P.; Yi, P.F.; Hu, R.S. Integrative analysis of transcriptome and metabolome provides insights into the underlying mechanism of cold stress response and recovery in two tobacco cultivars. Environ. Exp. Bot. 2022, 200, 104920. [Google Scholar] [CrossRef]
- Zhou, L.X.; Yarra, R.; Jin, L.F.; Cao, H.X. Genome-wide identification and expression analysis of MYB gene family in oil palm (Elaeis guineensis Jacq.) under abiotic stress conditions. Environ. Exp. Bot. 2020, 180, 104245. [Google Scholar] [CrossRef]
- Zhou, L.X.; Yarra, R.; Yang, Y.D.; Liu, Y.J.; Yang, M.D.; Cao, H.X. The oil palm R2R3-MYB subfamily genes EgMYB111 and EgMYB157 improve multiple abiotic stress tolerance in transgenic Arabidopsis plants. Plant Cell Rep. 2022, 41, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Saand, M.A.; Li, J.; Wu, Y.; Zhou, L.; Cao, H.; Yang, Y. Integrative Omics Analysis of Three Oil Palm Varieties Reveals (Tanzania × Ekona) TE as a Cold-Resistant Variety in Response to Low-Temperature Stress. Int. J. Mol. Sci. 2022, 23, 14926. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.X.; Lei, X.T.; Liu, Y.J.; Sun, C.X.; Zhang, R.L. Identification and comprehensive evaluation of cold resistance indexes of coconut. Guangdong Agric. Sci. 2016, 2, 49–54. (In Chinese) [Google Scholar]
- Yang, Y.D.; Mumtaz, A.S.; Walid, B.A.; Zhang, J.; Wu, Y.; Li, J.; Fan, H.K.; Wang, F.Y. iTRAQ-based comparative proteomic analysis of two coconut varieties reveals aromatic coconut cold-sensitive in response to low temperature. J. Proteom. 2020, 220, 103766. [Google Scholar] [CrossRef]
- Abdel-Moneim, A.M.; Al-Kahtani, M.A.; El-Kersh, M.A.; Al-Omair, M.A. Free radical-scavenging, anti-inflammatory/anti-fibrotic and hepatoprotective actions of taurine and silymarin against CCl4 induced rat liver damage. PLoS ONE 2015, 10, e0144509. [Google Scholar] [CrossRef]
- Sivankalyani, V.; Sela, N.; Feygenberg, O.; Zemach, H.; Maurer, D.; Alkan, N. Transcriptome dynamics in mango fruit peel reveals mechanisms of chilling stress. Front. Plant Sci. 2016, 7, 1579. [Google Scholar] [CrossRef]
- Tiwari, S.; Patel, A.; Singh, M.; Prasad, S.M. Regulation of temperature stress in plants. In Plant Life under Changing Environment; Tripathi, D.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 25–45. [Google Scholar] [CrossRef]
- Ahmad, P.; Abd_Allah, E.F.; Alyemeni, M.N.; Wijaya, L.; Alam, P.; Bhardwaj, R.; Siddique, H.M.K. Exogenous application of calcium to 24-epibrassinosteroid pre-treated tomato seedlings mitigates NaCl toxicity by modifying ascorbate-glutathione cycle and secondary metabolites. Sci. Rep. 2018, 8, 13515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xiang, X.; Liu, D.; Yang, A.; Wang, Y. Tobacco transcription factor NtbHLH123 confers tolerance to cold stress by regulating the NtCBF pathway and reactive oxygen species homeostasis. Front. Plant Sci. 2018, 9, 381. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef]
- Meloni, D.A.; Oliva, M.A.; Martinez, C.A.; Cambraia, J. Photosynthesis and activity of superoxide dismutase, peroxidase and glutathione reductase in cotton under salt stress. Environ. Exp. Bot. 2003, 49, 69–76. [Google Scholar] [CrossRef]
- Ashraf, M.F.M.R.; Foolad, M.R. Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ. Exp. Bot. 2007, 59, 206–216. [Google Scholar] [CrossRef]
- Wang, Y.X.; Hu, Y.; Chen, B.H.; Zhu, Y.F.; Dawuda, M.M.; Svetla, S. Physiological mechanisms of resistance to cold stress associated with 10 elite apple rootstocks. J. Integr. Agric. 2018, 17, 857–866. [Google Scholar] [CrossRef]
- Song, Y.P.; Ci, D.; Tian, M.; Zhang, D.Q. Comparison of the physiological effects and transcriptome responses of Populus simonii under different abiotic stresses. Plant Mol. Biol. 2014, 86, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Sazegari, S.; Niazi, A.; Ahmadi, F.S. A study on the regulatory network with promoter analysis for Arabidopsis DREB-genes. Bioinformation 2015, 11, 101. [Google Scholar] [CrossRef]
- Li, Y.Y.; Wang, X.W.; Ban, Q.Y.; Zhu, X.X.; Jiang, C.J.; Wei, C.L.; Bennetzen, J.L. Comparative transcriptomic analysis reveals gene expression associated with cold adaptation in the tea plant Camellia sinensis. BMC Genom. 2019, 20, 624. [Google Scholar] [CrossRef]
- Vazquez-Hernandez, M.; Romero, I.; Escribano, M.I.; Merodio, C.; Sanchez-Ballesta, M.T. Deciphering the Role of CBF/DREB Transcription Factorsand Dehydrins in Maintaining the Quality of Table Grapes cv. Autumn Royal Treated with High CO2 Levels and Stored at 0 °C. Front. Plant Sci. 2017, 8, 1591. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.; Zhang, J.; Chen, J.; Wu, T.; Zhu, S.; Zhong, G. Expressing a Citrus ortholog of Arabidopsis ERF1 enhanced cold-tolerance in tobacco. Sci. Hortic. 2014, 174, 65–76. [Google Scholar] [CrossRef]
- Yun, K.Y.; Park, M.R.; Mohanty, B.; Herath, V.; Xu, F.; Mauleon, R.; Wijaya, E.; Bajic, V.B.; Bruskiewich, R.; Reyes, B.G. Transcriptional regulatory network triggered by oxidative signals configures the early response mechanisms of japonica rice to chilling stress. BMC Plant Biol. 2010, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 86–96. [Google Scholar] [CrossRef]
- Pradhan, S.K.; Pandit, E.; Nayak, D.K.; Behera, L.; Mohapatra, T. Genes, pathways and transcription factors involved in seedling stage chilling stress tolerance in indica rice through RNA-Seq analysis. BMC Plant Biol. 2019, 19, 352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, T.; Sun, X.; Wang, Y.; Du, C.; Zhu, Z.; Gichuki, D.K.; Wang, Q.F.; Li, S.H.; Xin, H.P. Overexpression of VaWRKY12, a transcription factor from Vitis amurensis with increased nuclear localization under low temperature, enhances cold tolerance of plants. Plant Mol. Biol. 2019, 100, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jia, J.; Zhao, P.; Guo, X.; Chen, S.; Qi, D.; Cheng, L.; Liu, G. LcMYB4, an unknown function transcription factor gene from sheepgrass, as a positive regulator of chilling and freezing tolerance in transgenic Arabidopsis. BMC Plant Biol. 2020, 20, 238. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Liu, H.C.; Zhang, X.S.; Guo, Q.X.; Bian, S.M.; Wang, J.Y.; Zhai, L.L. VcMYB4a, an R2R3-MYB transcription factor from Vaccinium corymbosum, negatively regulates salt, drought, and temperature stress. Gene 2020, 757, 144935. [Google Scholar] [CrossRef] [PubMed]
- Olate, E.; Jiménez-Gómez, J.M.; Holuigue, L.; Salinas, J. NPR1 mediates a novel regulatory pathway in cold acclimation by interacting with HSFA1 factors. Nat. Plants 2018, 4, 811–823. [Google Scholar] [CrossRef]
- Lang, S.; Liu, X.; Xue, H.; Li, X.; Wang, X. Functional characterization of BnHSFA4a as a heat shock transcription factor in controlling the reestablishment of desiccation tolerance in seeds. J. Exp. Bot. 2017, 68, 2361–2375. [Google Scholar] [CrossRef]
- Wang, Y.J.; Zhang, Z.G.; He, X.J.; Zhou, H.L.; Wen, Y.X.; Dai, J.X.; Zhang, J.S.; Chen, S.Y. A rice transcription factor OsbHLH1 is involved in cold stress response. Theor. Appl. Genet. 2003, 107, 1402–1409. [Google Scholar] [CrossRef]
- Wang, D.Z.; Jin, Y.N.; Ding, X.H.; Wang, W.J.; Zhai, S.S.; Bai, L.P.; Guo, Z.F. Gene regulation and signal transduction in the ICE–CBF–COR signaling pathway during cold stress in plants. Biochemistry 2017, 82, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Lehti-Shiu, M.D.; Panchy, N.; Wang, P.; Uygun, S.; Shiu, S.H. Diversity, expansion, and evolutionary novelty of plant DNA-binding transcription factor families. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, S.J.; Sebolt, A.M.; Salazar, M.P.; Everard, J.D.; Thomashow, M.F. Overexpression of the Arabidopsis CBF3 transcriptional activator mimics multiple biochemical changes associated with cold acclimation. Plant Physiol. 2000, 124, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- He, W.D.; Gao, J.; Dou, T.X.; Shao, X.H.; Bi, F.C.; Sheng, O.; Deng, G.M.; Li, C.Y.; Hu, C.H.; Liu, J.H.; et al. Early Cold-Induced Peroxidases and Aquaporins Are Associated With High Cold Tolerance in Dajiao (Musa spp. ‘Dajiao’). Front. Plant Sci. 2018, 9, 282. [Google Scholar] [CrossRef]
- Rosa, P.; Antonio, B.; Roc, R.; Ramón, S.; José, M.M.S. BvCOLD1: A novel aquaporin from sugar beet (Beta vulgaris L.) involved in boron homeostasis and abiotic stress. Plant Cell Environ. 2018, 41, 2844–2857. [Google Scholar] [CrossRef]
- Janská, A.; Maršík, P.; Zelenková, S.; Ovesná, J. Cold stress and acclimation–what is important for metabolic adjustment? Plant Biol. 2010, 12, 395–405. [Google Scholar] [CrossRef]
- Carpaneto, A.; Ivashikina, N.; Levchenko, V.; Krol, E.; Jeworutzki, E.; Zhu, J.K.; Hedrich, R. Cold transiently activates calcium-permeable channels in Arabidopsis mesophyll cells. Plant Physiol. 2007, 143, 487–494. [Google Scholar] [CrossRef]
- Defalco, T.A.; Bender, K.W.; Snedden, W.A. Breaking the code: Ca2+ sensors in plant signalling. Biochem. J. 2010, 425, 27–40. [Google Scholar] [CrossRef]
- Guo, X.Y.; Liu, D.F.; Chong, K. Cold signaling in plants: Insights into mechanisms and regulation. J. Integr. Plant Biol. 2018, 60, 745–756. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, C.; Zhu, Y.; Zhang, L.; Chen, T.; Zhou, F.; Chen, H.; Lin, Y. The calcium-dependent kinase OsCPK24 functions in cold stress responses in rice. J. Integr. Plant Biol. 2018, 60, 173–188. [Google Scholar] [CrossRef]
- Rodriguez, M.C.; Petersen, M.; Mundy, J. Mitogen-activated protein kinase signaling in plants. Annu. Rev. Plant Biol. 2010, 61, 621–649. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ding, Y.; Yang, Y.; Song, C.; Wang, B.; Yang, S.; Guo, Y.; Gong, Z. Protein kinases in plant responses to drought, salt, and cold stress. J. Integr. Plant Biol. 2021, 63, 53–78. [Google Scholar] [CrossRef]
- Teige, M.; Scheikl, E.; Eulgem, T.; Dóczi, R.; Ichimura, K.; Shinozaki, K.; Dangl, J.L.; Hirt, H. The MKK2 pathway mediates cold and salt stress signaling in Arabidopsis. Mol. Cell 2004, 15, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Shad Ali, G.; Yang, L.; Du, L.; Reddy, A.S.; Poovaiah, B.W. Calcium/calmodulin regulated receptor-like kinase CRLK1 interacts with MEKK1 in plants. Plant Signal Behav. 2010, 5, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Bulgakov, V.P.; Wu, H.C.; Jinn, T.L. Coordination of ABA and chaperone signaling in plant stress responses. Trends Plant Sci. 2019, 24, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.L.; Yan, J.Y.; Su, Z.Z.; Chang, J.J.; Yang, J.Q.; Wei, C.H.; Zhang, Y.; Ma, J.X.; Zhang, X.; Li, H. Abscisic acid mediates grafting-induced cold tolerance of watermelon via interaction with melatonin and methyl jasmonate. Front. Plant Sci. 2021, 12, 785317. [Google Scholar] [CrossRef]
- Shi, Y.T.; Yang, S.H. ABA regulation of the cold stress response in plants. In Abscisic Acid: Metabolism, Transport and Signaling; Zhang, D.P., Ed.; Springer: Dordrecht, The Netherlands, 2014; pp. 337–363. [Google Scholar] [CrossRef]
- Hu, W.; Ding, Z.H.; Tie, W.W.; Yan, Y.; Liu, Y.; Wu, C.L.; Liu, J.H.; Wang, J.S.; Peng, M.; Xu, B.Y.; et al. Comparative physiological and transcriptomic analyses provide integrated insight into osmotic, cold, and salt stress tolerance mechanisms in banana. Sci. Rep. 2017, 7, 43007. [Google Scholar] [CrossRef]
- Tian, J.Y.; Ma, Y.; Tian, L.; Huang, C.; Chen, M.; Wei, A.Z. Comparative physiology and transcriptome response patterns in cold-tolerant and cold-sensitive varieties of Zanthoxylum bungeanum Maxim. Ind. Crop. Prod. 2021, 167, 113562. [Google Scholar] [CrossRef]
- Lumba, S.; Toh, S.; Handfield, L.F.; Swan, M.; Liu, R.; Youn, J.Y.; Cutler, S.R.; Subramaniam, R.; Provart, N.; Moses, A.; et al. A mesoscale abscisic acid hormone interactome reveals a dynamic signaling landscape in Arabidopsis. Dev. Cell. 2014, 29, 360–372. [Google Scholar] [CrossRef]
- Sah, S.K.; Reddy, K.R.; Li, J.X. Abscisic acid and abiotic stress tolerance in crop plants. Front. Plant Sci. 2016, 7, 571. [Google Scholar] [CrossRef]
- Paterlini, A. Uncharted routes: Exploring the relevance of auxin movement via plasmodesmata. Biol. Open. 2020, 9, bio055541. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K.; Uemura, M.; Tsurumi, S.; Rahman, A. Auxin response in Arabidopsis under cold stress: Underlying molecular mechanisms. Plant Cell. 2009, 21, 3823–3838. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Hagen, B.G.; Guilfoyle, T.J. The roles of auxin response factor domains in auxin-responsive transcription. Plant Cell. 2003, 15, 533–543. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Lin, Y.; Luo, Y.; Wang, X.; Chen, Q.; Sun, B.; Wang, Y.; Li, M.; Tang, H. A transcriptomic analysis reveals diverse regulatory networks that respond to cold stress in strawberry (Fragaria × ananassa). Int. J. Genom. 2019, 2019, 7106092. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; Zhang, J.; Wang, S.J.; Zhang, H.E.; Liu, Y.C.; Yang, M.S. Integrative transcriptomic and metabolomic analyses reveal the flavonoid biosynthesis of Pyrus hopeiensis flowers under cold stress. Hortic. Plant J. 2023, 9, 395–413. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, D.; Zhou, R.; Wang, X.; Dossa, K.; Wang, L.; Zhang, Y.; Yu, J.; Gong, H.; Zhang, X. Transcriptome and metabolome analyses of two contrasting sesame genotypes reveal the crucial biological pathways involved in rapid adaptive response to salt stress. BMC Plant Biol. 2019, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Lunn, J.E.; Delorge, I.; Figueroa, C.M.; Van Dijck, P.; Stitt, M. Trehalose metabolism in plants. Plant J. 2014, 79, 544–567. [Google Scholar] [CrossRef]
- Ma, S.; Lv, J.; Li, X.; Ji, T.; Zhang, Z.; Gao, L. Galactinol synthase gene 4 (CsGolS4) increases cold and drought tolerance in Cucumis sativus L. by inducing RFO accumulation and ROS scavenging. Environ. Exp. Bot. 2021, 185, 104406. [Google Scholar] [CrossRef]
- Matros, A.; Peshev, D.; Peukert, M.; Mock, H.P.; Van Den Ende, W. Sugars as hydroxyl radical scavengers: Proof-of-concept by studying the fate of sucralose in Arabidopsis. Plant J. 2015, 82, 822–839. [Google Scholar] [CrossRef]
- Sengupta, S.; Mukherjee, S.; Basak, P.; Majumder, A.L. Significance of galactinol and raffinose family oligosaccharide synthesis in plants. Front. Plant Sci. 2015, 6, 656. [Google Scholar] [CrossRef]
- Wei, L.; Sun, X.; Qi, X.; Zhang, Y.; Xu, Y. Dihydromyricetin ameliorates cardiac ischemia/reperfusion injury through Sirt3 activation. BioMed. Res. Int. 2019, 2019, 6803943. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tikunov, Y.; Schouten, R.E.; Marcelis, L.F.M.; Visser, R.G.F.; Bovy, A. Anthocyanin biosynthesis and degradation mechanisms in solanaceous vegetables: A review. Front. Chem. 2018, 6, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.L.; Kang, S.M.; Dhakal, K.H.; Hussain, J.; Adnan, M.; Kim, J.G.; Lee, I.J. Flavonoids and amino acid regulation in Capsicum annuum L. by endophytic fungi under different heat stress regimes. Sci. Hortic. 2013, 155, 1–7. [Google Scholar] [CrossRef]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Zhang, J.; Yin, X.R.; Li, H.; Xu, M.; Zhang, M.X.; Li, S.J.; Liu, X.F.; Shi, Y.N.; Grierson, D.; Chen, K.S. Ethylene response factor EjERF39- EjMYB8 complex activates cold-induced lignification of loquat fruit, via the biosynthetic gene Ej4CL1. J. Exp. Bot. 2020, 71, 3172–3184. [Google Scholar] [CrossRef]
- An, J.P.; Wang, X.F.; Zhang, X.W.; Xu, H.F.; Hao, Y.J. An apple MYB transcription factor regulates cold tolerance and anthocyanin accumulation and undergoes MIEL1-mediated degradation. Plant Biotechnol. J. 2020, 18, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S. Principles and Techniques of Plant Physiology and Biochemistry Experiment; Higher Education Press: Beijing, China, 2000; pp. 25–63. [Google Scholar]
- Lu, L.; Chen, S.; Yang, W.; Wu, Y.; Liu, Y.; Yin, X.; Yang, Y.; Yang, Y. Integrated transcriptomic and metabolomic analyses reveal key metabolic pathways in response to potassium deficiency in coconut (Cocos nucifera L.)seedlings. Front. Plant Sci. 2023, 14, 1112264. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Xiao, Y.; Xu, P.X.; Fan, H.K.; Luc, B.; Xia, W.; Stephanie, B.; Xu, J.Y.; Li, Q.; Guo, A.P.; Zhou, L.X.; et al. The genome draft of coconut (Cocos nucifera). GigaScience 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology; R package Version 2.36.0. 2019. Available online: http://doi.org/10.18129/B9.bioc.topGO (accessed on 1 August 2023).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Zhang, G.; He, P.; Tan, H.; Budhu, A.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.G.; Lee, D.H.; Maitra, A.; et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin. Cancer Res. 2013, 15, 4983–4993. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, L.; Yang, W.; Dong, Z.; Tang, L.; Liu, Y.; Xie, S.; Yang, Y. Integrated Transcriptomic and Metabolomics Analyses Reveal Molecular Responses to Cold Stress in Coconut (Cocos nucifera L.) Seedlings. Int. J. Mol. Sci. 2023, 24, 14563. https://doi.org/10.3390/ijms241914563
Lu L, Yang W, Dong Z, Tang L, Liu Y, Xie S, Yang Y. Integrated Transcriptomic and Metabolomics Analyses Reveal Molecular Responses to Cold Stress in Coconut (Cocos nucifera L.) Seedlings. International Journal of Molecular Sciences. 2023; 24(19):14563. https://doi.org/10.3390/ijms241914563
Chicago/Turabian StyleLu, Lilan, Weibo Yang, Zhiguo Dong, Longxiang Tang, Yingying Liu, Shuyun Xie, and Yaodong Yang. 2023. "Integrated Transcriptomic and Metabolomics Analyses Reveal Molecular Responses to Cold Stress in Coconut (Cocos nucifera L.) Seedlings" International Journal of Molecular Sciences 24, no. 19: 14563. https://doi.org/10.3390/ijms241914563