
Molecular Mechanism of STIL Coiled-Coil Domain Oligomerization

Abstract
:1. Introduction
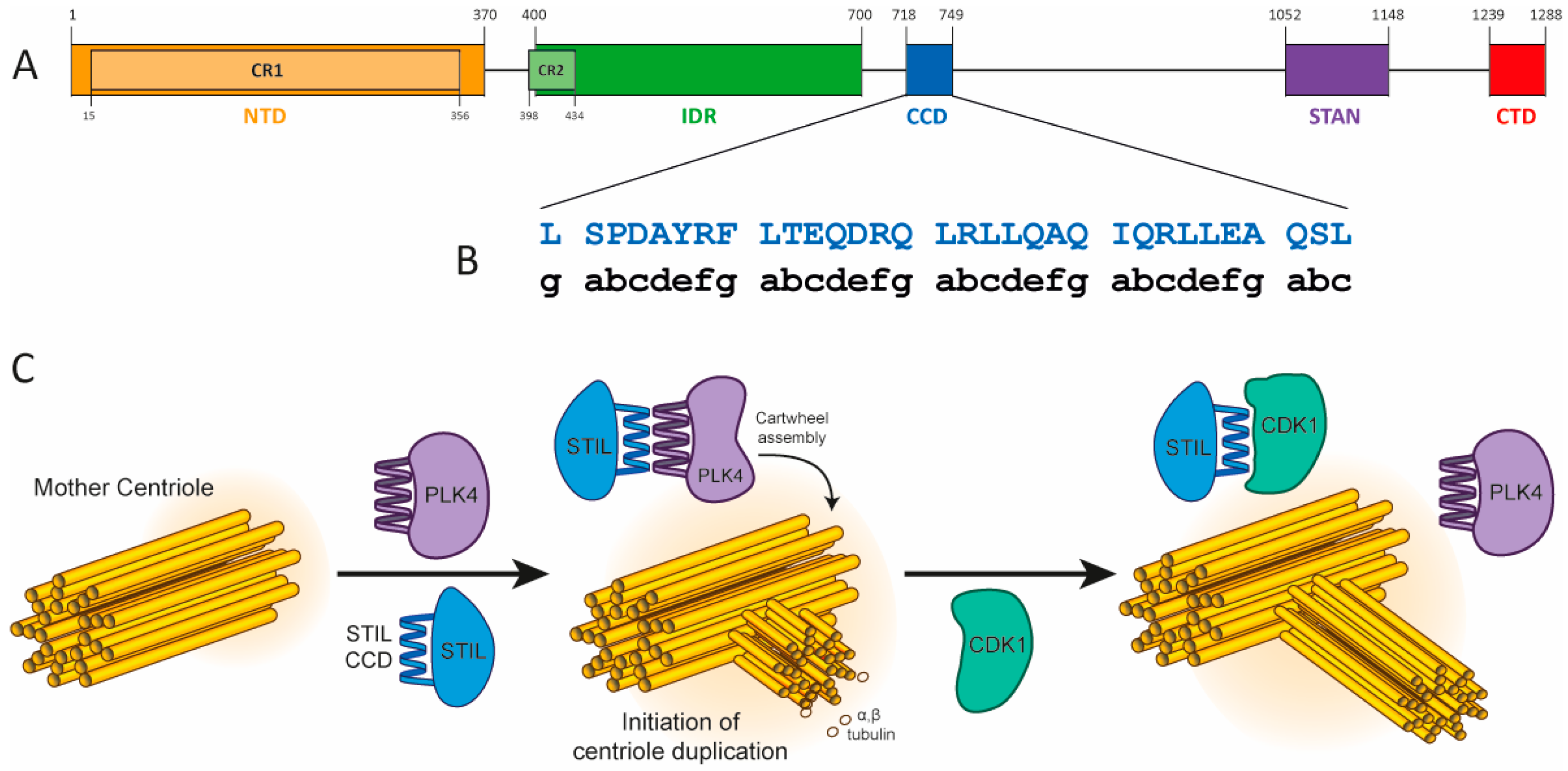
2. Results
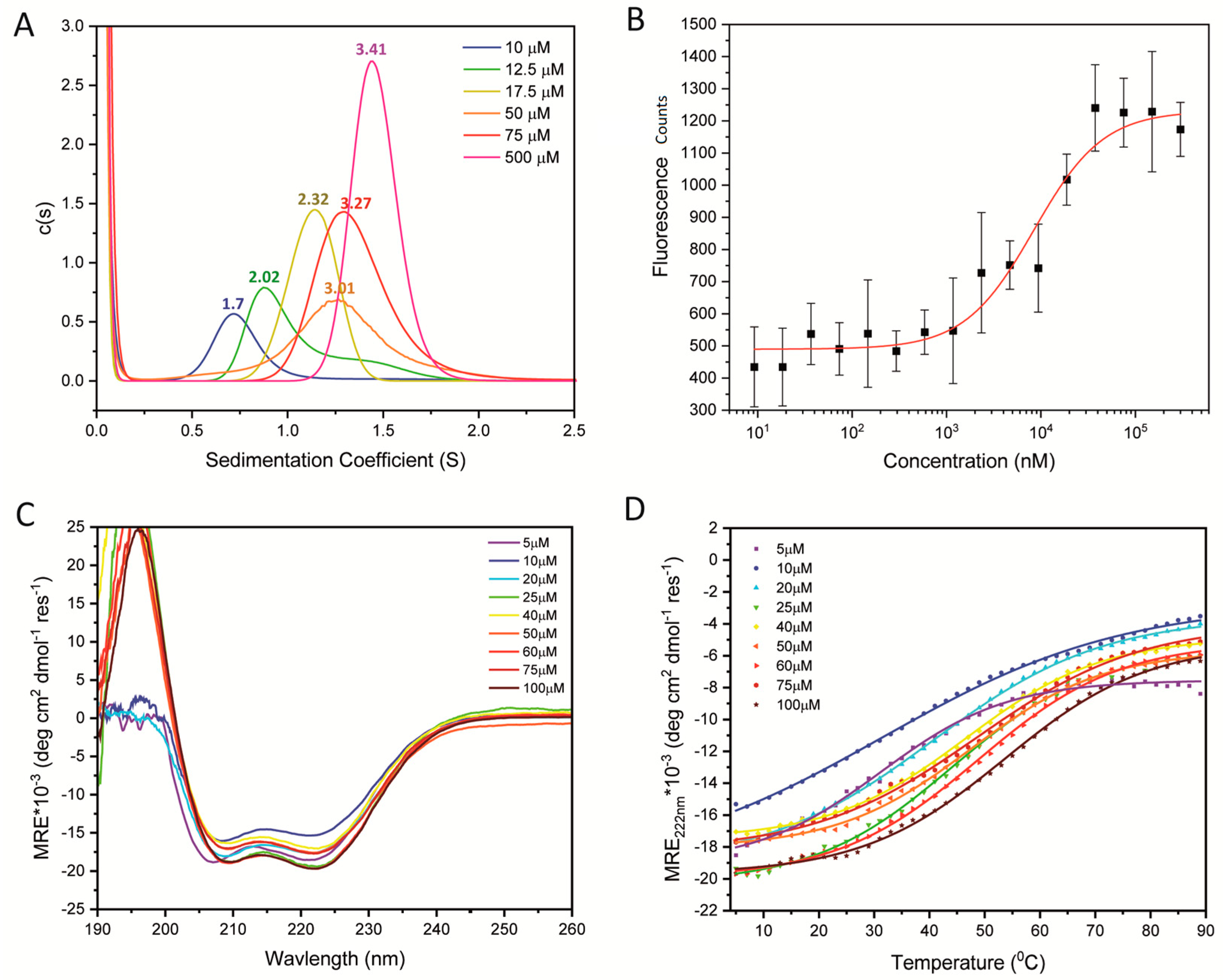
2.1. Quantifying the Oligomerization of WT STIL CCD Peptide
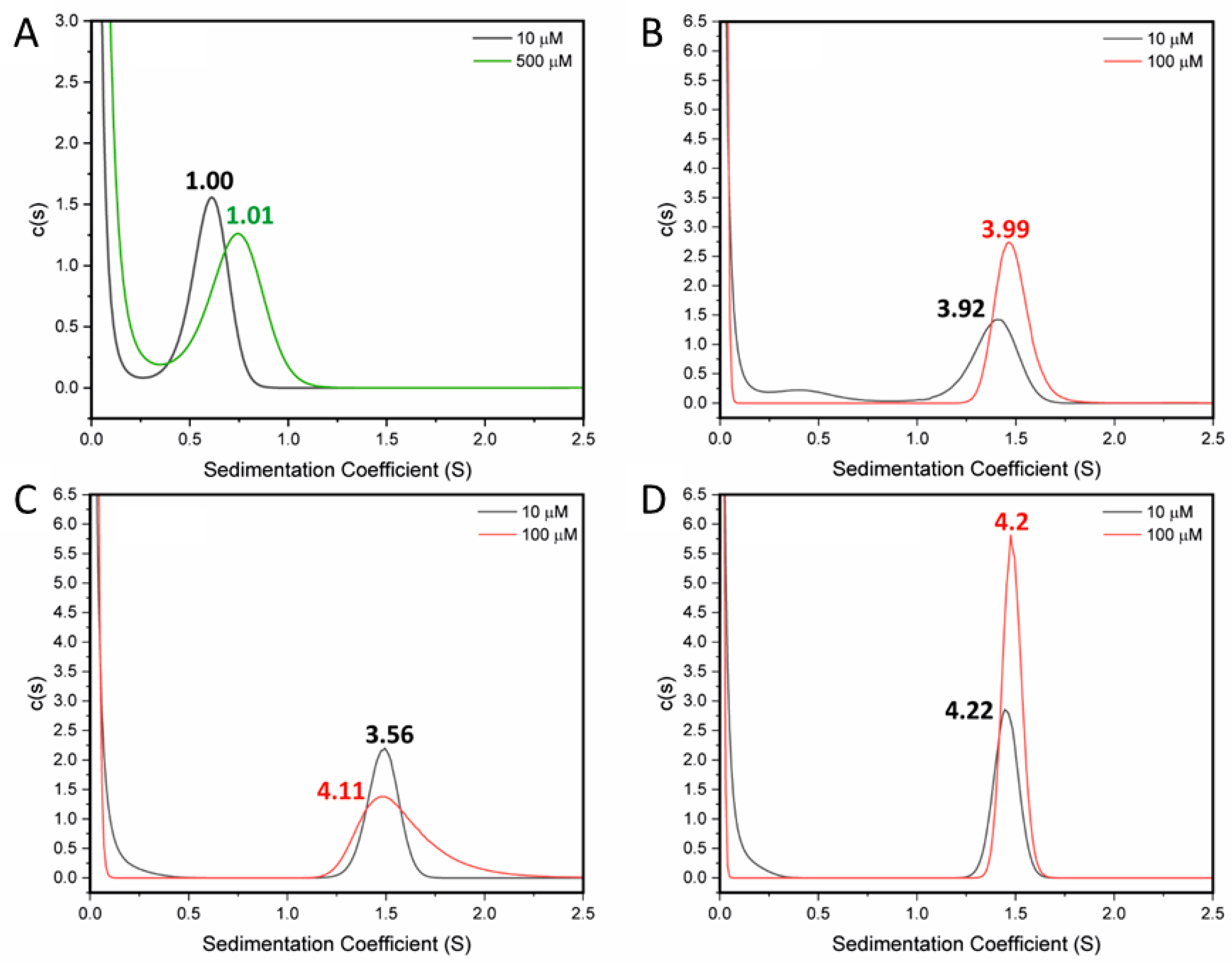
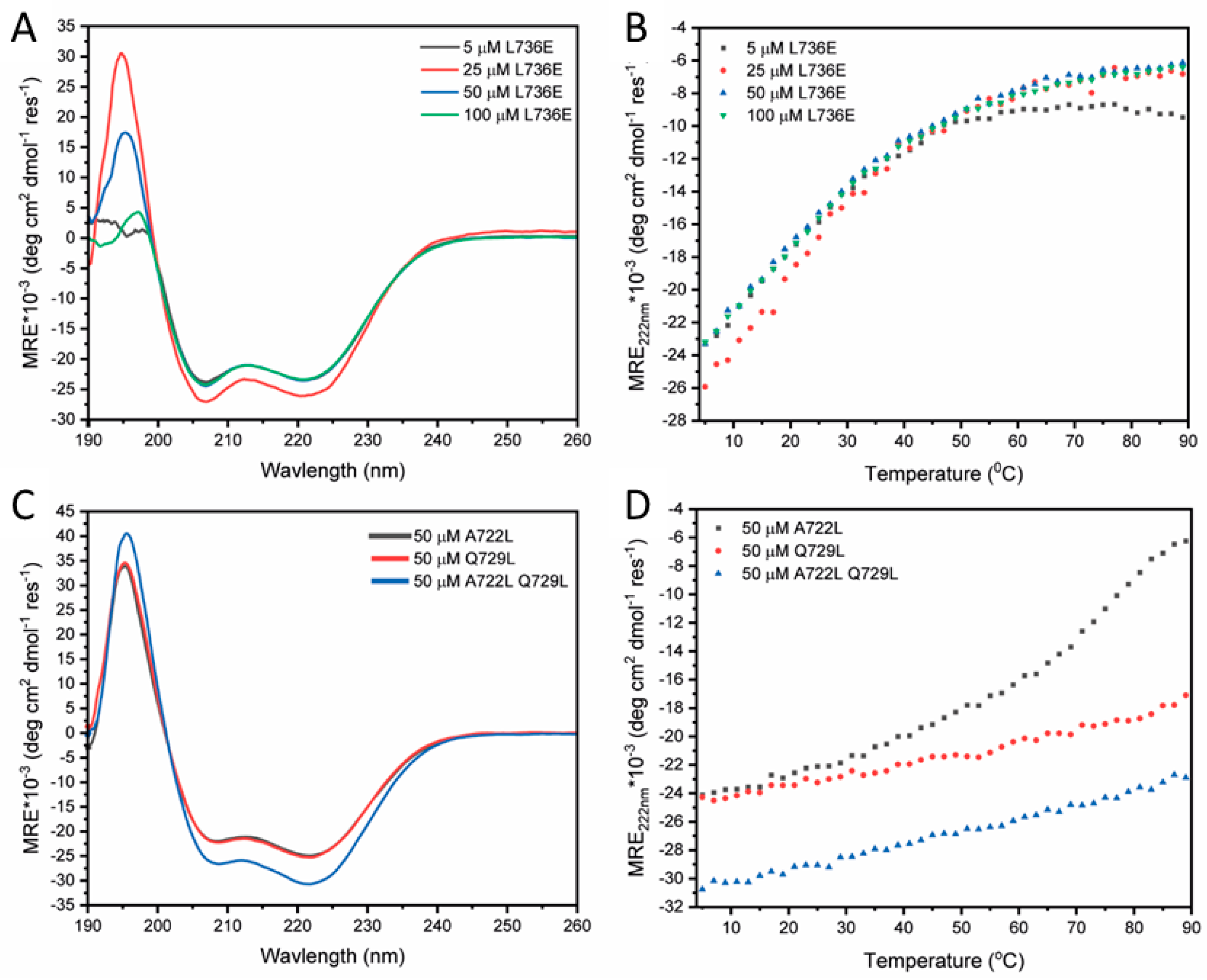
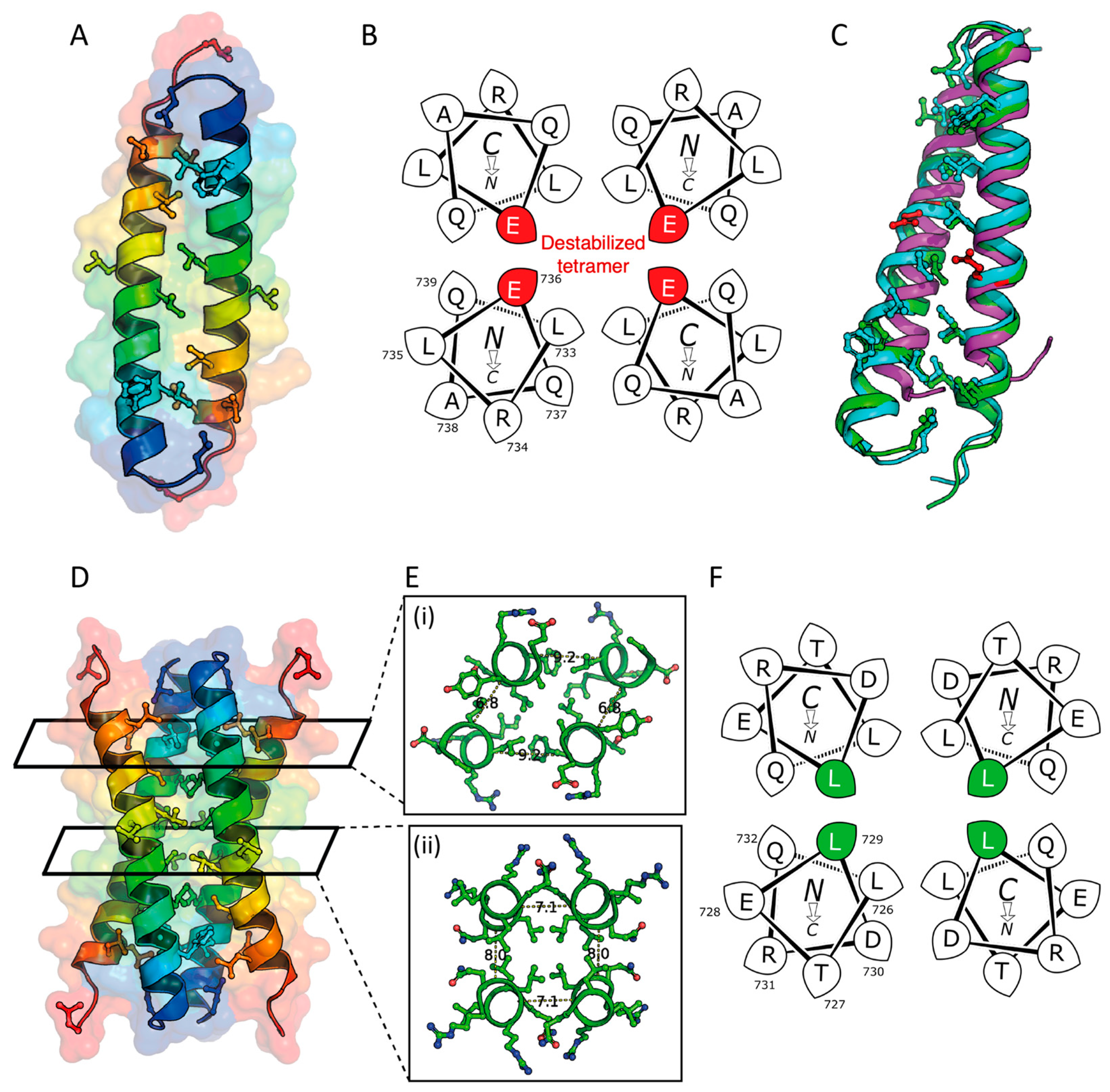
2.2. Destabilising and Stabilising Mutations in the CCD Peptide of STIL
2.3. X-ray Crystal Structures of the L736E and Q729L Variants
3. Discussion
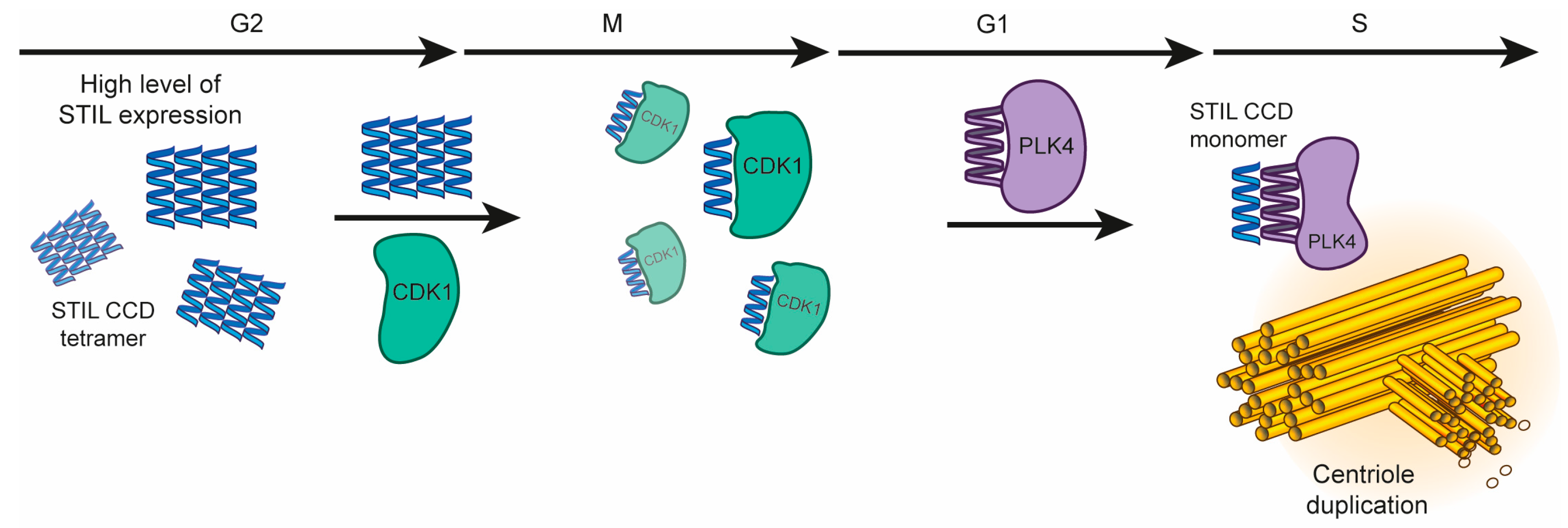
3.1. The Role of STIL CCD Oligomerization during Centriole Duplication
3.2. Destabilizing and Stabilizing Mutants of STIL CCD Peptide Leads to Monomeric and Tetrameric Oligomeric States, Respectively
3.3. STIL CCD Activity during Centriole Duplication
4. Materials and Methods
4.1. Peptide Synthesis, Labelling and Purification
4.2. Crystallization
4.3. AUC
4.4. CD Spectroscopy
4.5. Fluorescence
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Izraeli, S.; Colaizzo-Anas, T.; Bertness, V.L.; Mani, K.; Aplan, P.D.; Kirsch, I.R. Expression of the SIL Gene Is Correlated with Growth Induction and Cellular Proliferation. Cell Growth Differ. 1997, 8, 1171–1179. [Google Scholar] [PubMed]
- Castiel, A.; Danieli, M.M.; David, A.; Moshkovitz, S.; Aplan, P.D.; Kirsch, I.R.; Brandeis, M.; Krämer, A.; Izraeli, S. The Stil Protein Regulates Centrosome Integrity and Mitosis through Suppression of Chfr. J. Cell Sci. 2011, 124, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Arquint, C.; Nigg, E.A. STIL Microcephaly Mutations Interfere with APC/C-Mediated Degradation and Cause Centriole Amplification. Curr. Biol. 2014, 24, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Rabinowicz, N.; Mangala, L.S.; Brown, K.R.; Checa-Rodriguez, C.; Castiel, A.; Moskovich, O.; Zarfati, G.; Trakhtenbrot, L.; Levy-Barda, A.; Jiang, D.; et al. Targeting the Centriolar Replication Factor STIL Synergizes with DNA Damaging Agents for Treatment of Ovarian Cancer. Oncotarget 2017, 8, 27380–27392. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A Molecular Signature of Metastasis in Primary Solid Tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Kratz, A.-S.; Barenz, F.; Richter, K.T.; Hoffmann, I. Plk4-Dependent Phosphorylation of STIL Is Required for Centriole Duplication. Biol. Open 2015, 4, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Moyer, T.C.; Clutario, K.M.; Lambrus, B.G.; Daggubati, V.; Holland, A.J. Binding of STIL to Plk4 Activates Kinase Activity to Promote Centriole Assembly. J. Cell Biol. 2015, 209, 863–878. [Google Scholar] [CrossRef]
- Vulprecht, J.; David, A.; Tibelius, A.; Castiel, A.; Konotop, G.; Liu, F.; Bestvater, F.; Raab, M.S.; Zentgraf, H.; Izraeli, S.; et al. STIL Is Required for Centriole Duplication in Human Cells. J. Cell Sci. 2012, 125, 1353–1362. [Google Scholar] [CrossRef]
- Arquint, C.; Gabryjonczyk, A.M.; Imseng, S.; Böhm, R.; Sauer, E.; Hiller, S.; Nigg, E.A.; Maier, T. STIL Binding to Polo-Box 3 of PLK4 Regulates Centriole Duplication. eLife 2015, 4, 7554. [Google Scholar] [CrossRef]
- Zitouni, S.; Francia, M.E.; Leal, F.; Montenegro Gouveia, S.; Nabais, C.; Duarte, P.; Gilberto, S.; Brito, D.; Moyer, T.; Kandels-Lewis, S.; et al. CDK1 Prevents Unscheduled PLK4-STIL Complex Assembly in Centriole Biogenesis. Curr. Biol. 2016, 26, 1127–1137. [Google Scholar] [CrossRef]
- Pihan, G.A. Centrosome Dysfunction Contributes to Chromosome Instability, Chromoanagenesis, and Genome Reprograming in Cancer. Front. Oncol. 2013, 3, 277. [Google Scholar] [CrossRef] [PubMed]
- Dhruti, P.; Shyamala, M.; Sandrine, P.; Pierre, G.; Vincent, E.G. STIL Balancing Primary Microcephaly and Cancer Review-Article. Cell Death Dis. 2018, 9, 65. [Google Scholar] [CrossRef]
- Cottee, M.A.; Muschalik, N.; Johnson, S.; Leveson, J.; Raff, J.W.; Lea, S.M. The Homo-Oligomerisation of Both Sas-6 and Ana2 Is Required for Efficient Centriole Assembly in Flies. eLife 2015, 4, e07236. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Amartely, H.; Rabinowicz, N.; Shamir, M.; Friedler, A.; Izraeli, S. Molecular Basis of the STIL Coiled Coil Oligomerization Explains Its Requirement for de-Novo Formation of Centrosomes in Mammalian Cells. Sci. Rep. 2016, 6, 24296. [Google Scholar] [CrossRef]
- Cottee, M.A.; Johnson, S.; Raff, J.W.; Lea, S.M. A Key Centriole Assembly Interaction Interface between Human PLK4 and STIL Appears to Not Be Conserved in Flies. Biol. Open 2017, 6, 381–389. [Google Scholar] [CrossRef]
- Amartely, H.; David, A.; Shamir, M.; Lebendiker, M.; Izraeli, S.; Friedler, A.; Arias-Moreno, X. Differential Effects of Zinc Binding on Structured and Disordered Regions in the Multidomain STIL Protein. Chem. Sci. 2016, 7, 4140–4147. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.; Schraegle, S.J.; Stahlberg, E.A.; Meier, I. Coiled-Coil Protein Composition of 22 Proteomes—Differences and Common Themes in Subcellular Infrastructure and Traffic Control. BMC Evol. Biol. 2005, 5, 66. [Google Scholar] [CrossRef]
- Rackham, O.J.L.; Madera, M.; Armstrong, C.T.; Vincent, T.L.; Woolfson, D.N.; Gough, J. The Evolution and Structure Prediction of Coiled Coils across All Genomes. J. Mol. Biol. 2010, 403, 480–493. [Google Scholar] [CrossRef]
- Dixon, A.S.; Pendley, S.S.; Bruno, B.J.; Woessner, D.W.; Shimpi, A.A.; Cheatham, T.E.; Lim, C.S. Disruption of Bcr-Abl Coiled Coil Oligomerization by Design. J. Biol. Chem. 2011, 31, 27751–27760. [Google Scholar] [CrossRef]
- Lapenta, F.; Aupič, J.; Strmšek, Ž.; Jerala, R. Coiled Coil Protein Origami: From Modular Design Principles towards Biotechnological Applications. Chem. Soc. Rev. 2018, 47, 3530–3542. [Google Scholar] [CrossRef]
- Lupas, A.N.; Bassler, J. Coiled Coils—A Model System for the 21st Century. Trends Biochem. Sci. 2017, 42, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Packing of Alpha-Helices: Simple Coiled-Coils. Acta Crystallogr. 1953, 6, 986–997. [Google Scholar] [CrossRef]
- Kumar, P.; Woolfson, D.N. Socket2: A Program for Locating, Visualizing and Analyzing Coiled-Coil Interfaces in Protein Structures. Bioinformatics 2021, 37, 4575–4577. [Google Scholar] [CrossRef] [PubMed]
- Walshaw, J.; Woolfson, D.N. SOCKET: A Program for Identifying and Analysing Coiled-Coil Motifs within Protein Structures. J. Mol. Biol. 2001, 307, 1427–1450. [Google Scholar] [CrossRef] [PubMed]
- Woolfson, D.N. Coiled-Coil Design: Updated and Upgraded. Subcell. Biochem. 2017, 82, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A.N.; Bassler, J.; Dunin-Horkawicz, S. The Structure and Topology of α-Helical Coiled Coils. Subcell. Biochem. 2017, 82, 95–129. [Google Scholar] [CrossRef] [PubMed]
- Walshaw, J.; Woolfson, D.N. Extended Knobs-into-Holes Packing in Classical and Complex Coiled-Coil Assemblies. J. Struct. Biol. 2003, 3, 349–361. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Horner, K.A.; Bartlett, G.J.; Rhys, G.G.; Wilson, A.J.; Woolfson, D.N. De Novo Coiled-Coil Peptides as Scaffolds for Disrupting Protein-Protein Interactions. Chem. Sci. 2018, 9, 7656–7665. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Boyle, A.L.; Bruning, M.; Bartlett, S.J.; Vincent, T.L.; Zaccai, N.R.; Armstrong, C.T.; Bromley, E.H.C.; Booth, P.J.; Brady, R.L.; et al. A Basis Set of de Novo Coiled-Coil Peptide Oligomers for Rational Protein Design and Synthetic Biology. ACS Synth. Biol. 2012, 1, 240–250. [Google Scholar] [CrossRef]
- Delorenzi, M.; Speed, T. An HMM Model for Coiled-Coil Domains and a Comparison with PSSM-Based Predictions. Bioinformatics 2002, 18, 617–625. [Google Scholar] [CrossRef]
- Rhys, G.G.; Wood, C.W.; Beesley, J.L.; Zaccai, N.R.; Burton, A.J.; Brady, R.L.; Thomson, A.R.; Woolfson, D.N. Navigating the Structural Landscape of de Novo α-Helical Bundles. J. Am. Chem. Soc. 2019, 141, 8787–8797. [Google Scholar] [CrossRef] [PubMed]
- Testa, O.D.; Moutevelis, E.; Woolfson, D.N. CC+: A Relational Database of Coiled-Coil Structures. Nucleic Acids Res. 2009, 37, D315–D322. [Google Scholar] [CrossRef] [PubMed]
- Millán, C.; Sammito, M.; Usón, I. Macromolecular Ab Initio Phasing Enforcing Secondary and Tertiary Structure. IUCrJ 2015, 2, 95–105. [Google Scholar] [CrossRef]
- Hatzopoulos, G.N.; Erat, M.C.; Cutts, E.; Rogala, K.B.; Slater, L.M.; Stansfeld, P.J.; Vakonakis, I. Structural Analysis of the G-Box Domain of the Microcephaly Protein CPAP Suggests a Role in Centriole Architecture. Structure 2013, 21, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Good, M.C.; Zalatan, J.G.; Lim, W.A. Scaffold Proteins: Hubs for Controlling the Flow of Cellular Information. Science 2011, 332, 680–686. [Google Scholar] [CrossRef]
- Garbett, D.; Bretscher, A. The Surprising Dynamics of Scaffolding Proteins. Mol. Biol. Cell 2014, 25, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Campaner, S.; Kaldis, P.; Izraeli, S.; Kirsch, I.R. Sil Phosphorylation in a Pin1 Binding Domain Affects the Duration of the Spindle Checkpoint. Mol. Cell. Biol. 2005, 25, 6660–6672. [Google Scholar] [CrossRef]
- Jancarik, J.; Kim, S.H. Sparse Matrix Sampling. A Screening Method for Crystallization of Proteins. J. Appl. Crystallogr. 1991, 24, 409–411. [Google Scholar] [CrossRef]
- Page, R.; Grzechnik, S.K.; Canaves, J.M.; Spraggon, G.; Kreusch, A.; Kuhn, P.; Stevens, R.C.; Lesley, S.A. Shotgun Crystallization Strategy for Structural Genomics: An Optimized Two-Tiered Crystallization Screen against the Thermotoga Maritima Proteome. Acta Crystallogr.—Sect. D Biol. Crystallogr. 2003, 59, 1028–1037. [Google Scholar] [CrossRef]
- Newman, J.; Egan, D.; Walter, T.S.; Meged, R.; Berry, I.; Ben Jelloul, M.; Sussman, J.L.; Stuart, D.I.; Perrakis, A. Towards Rationalization of Crystallization Screening for Small- To Medium-Sized Academic Laboratories: The PACT/JCSG+ Strategy. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 1426–1431. [Google Scholar] [CrossRef]
- Radaev, S.; Li, S.; Sun, P.D. A Survey of Protein-Protein Complex Crystallizations. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Radaev, S.; Sun, P.D. Crystallization of Protein-Protein Complexes. J. Appl. Crystallogr. 2002, 35, 674–676. [Google Scholar] [CrossRef]
- Gorrec, F. The MORPHEUS Protein Crystallization Screen. J. Appl. Crystallogr. 2009, 42, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and Evaluation of a New Integration Package. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Montero, L.; Parkhurst, J.; Gerstel, M.; Gildea, R.; Winter, G.; Vollmar, M.; Waterman, D.; Evans, G. Introducing DUI, a Graphical Interface for DIALS. Acta Crystallogr. Sect. A Found. Adv. 2016, 72, s189. [Google Scholar] [CrossRef]
- Potterton, L.; Agirre, J.; Ballard, C.; Cowtan, K.; Dodson, E.; Evans, P.R.; Jenkins, H.T.; Keegan, R.; Krissinel, E.; Stevenson, K.; et al. CCP 4 I 2: The New Graphical User Interface to the CCP 4 Program Suite. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 68–84. [Google Scholar] [CrossRef]
- Sammito, M.; Millán, C.; Rodríguez, D.D.; De Ilarduya, I.M.; Meindl, K.; De Marino, I.; Petrillo, G.; Buey, R.M.; De Pereda, J.M.; Zeth, K.; et al. Exploiting Tertiary Structure through Local Folds for Crystallographic Phasing. Nat. Methods 2013, 10, 1099–1104. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Cowtan, K. The Buccaneer Software for Automated Model Building. 1. Tracing Protein Chains. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 1002–1011. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Salzemann, J.; Bloch, V.; Stockinger, H.; Berglund, A.C.; Blanchet, C.; Bongcam-Rudloff, E.; Combet, C.; Da Costa, A.L.; Deleage, G.; et al. PDB-REDO: Automated Re-Refinement of X-Ray Structure Models in the PDB. J. Appl. Crystallogr. 2009, 42, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Cohn, E.; Edsall, J. Density and Apparent Specific Volume of Proteins; Proteins, Amino Acids and Peptides; ScienceOpen, Inc.: Boston, MA, USA, 1943. [Google Scholar]
- Demeler, B.; Gorbet, G.E. Analytical Ultracentrifugation Data Analysis with Ultrascan-III. In Analytical Ultracentrifugation: Instrumentation, Software, and Applications; Springer: Berlin/Heidelberg, Germany, 2016; pp. 119–143. ISBN 9784431559856. [Google Scholar]
- Zhao, H.; Piszczek, G.; Schuck, P. SEDPHAT—A Platform for Global ITC Analysis and Global Multi-Method Analysis of Molecular Interactions. Methods 2015, 76, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Thyparambil, A.A.; Latour, R.A. Protein Helical Structure Determination Using CD Spectroscopy for Solutions with Strong Background Absorbance from 190 to 230 Nm. Biochim. Biophys. Acta—Proteins Proteomics 2014, 1844, 2331–2337. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Amino Acid Sequence and CC Register | MRE222 nm (kdeg cm2 dmol−1 res−1) (% Helicity) * | Tm (°C) at 50 µM | AUC Oligomeric State (Starting Peptide Concentration(s)) | Oligomeric State (at 10 µM) | PDB ID | |
|---|---|---|---|---|---|---|---|
| g abcdefg abcdefg abcdefg abcdefg abc | SV (10 µM) | SE | |||||
| STIL CCD718–749 WT | L SPDAYRF LTEQDRQ LRLLQAQ IQRLLEA QSL-NH2 | −17.6 (47%) | 55.9 ± 0.5 | 1.7 | 2.9 ± 0.04 (5, 10, 20 µM) | Mixture of monomers and dimers | |
| STIL CCD718–749 L736E | L SPDAYRF LTEQDRQ LRLEQAQ IQRLLEA QSL-NH2 | −23.3 (62%) | 10 ± 2 | 1.00 | 0.79 ± 0.08 (500 µM) | Monomer | 8oyk |
| STIL CCD718–749 A722L | L SPDLYRF LTEQDRQ LRLLQAQ IQRLLEA QSL-NH2 | −24.9 (66%) | >70 | 3.92 | 3.51 ± 0.02 (5, 10, 20 µM) | Tetramer | |
| STIL CCD718–749 Q729L | L SPDAYRF LTELDRQ LRLLQAQ IQRLLEA QSL-NH2 | −25.3 (67%) | >95 | 3.56 | 3.47 ± 0.05 (5, 10, 20 µM) | Tetramer | 8oyl |
| STIL CCD718–749 A722L Q729L | L SPDLYRF LTELDRQ LRLLQAQ IQRLLEA QSL-NH2 | −30.7 (81%) | >95 | 4.22 | 3.24 ± 0.03 (5, 10, 20 µM) | Tetramer | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shamir, M.; Martin, F.J.O.; Woolfson, D.N.; Friedler, A. Molecular Mechanism of STIL Coiled-Coil Domain Oligomerization. Int. J. Mol. Sci. 2023, 24, 14616. https://doi.org/10.3390/ijms241914616
Shamir M, Martin FJO, Woolfson DN, Friedler A. Molecular Mechanism of STIL Coiled-Coil Domain Oligomerization. International Journal of Molecular Sciences. 2023; 24(19):14616. https://doi.org/10.3390/ijms241914616
Chicago/Turabian StyleShamir, Mai, Freddie J. O. Martin, Derek N. Woolfson, and Assaf Friedler. 2023. "Molecular Mechanism of STIL Coiled-Coil Domain Oligomerization" International Journal of Molecular Sciences 24, no. 19: 14616. https://doi.org/10.3390/ijms241914616