

Electrophoretic Determination of L-Carnosine in Health Supplements Using an Integrated Lab-on-a-Chip Platform with Contactless Conductivity Detection

 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
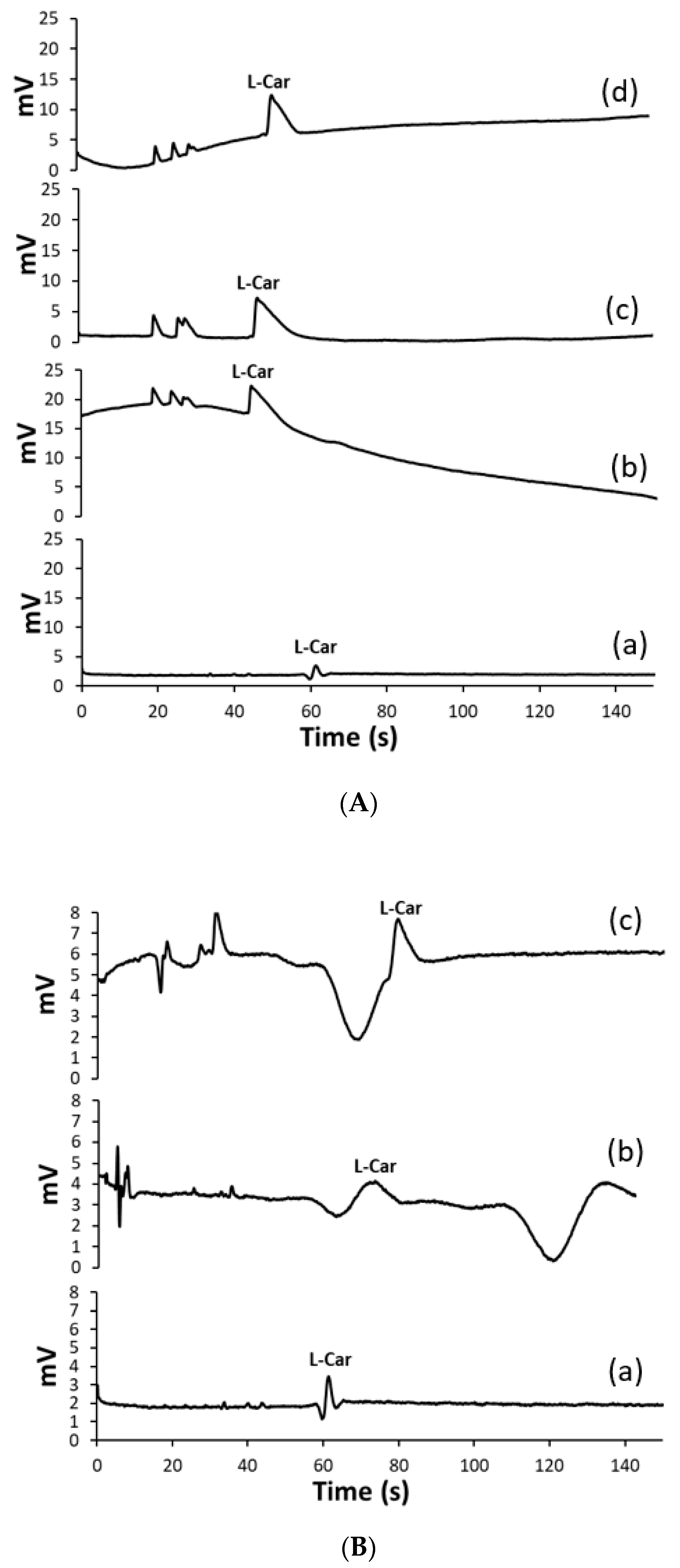
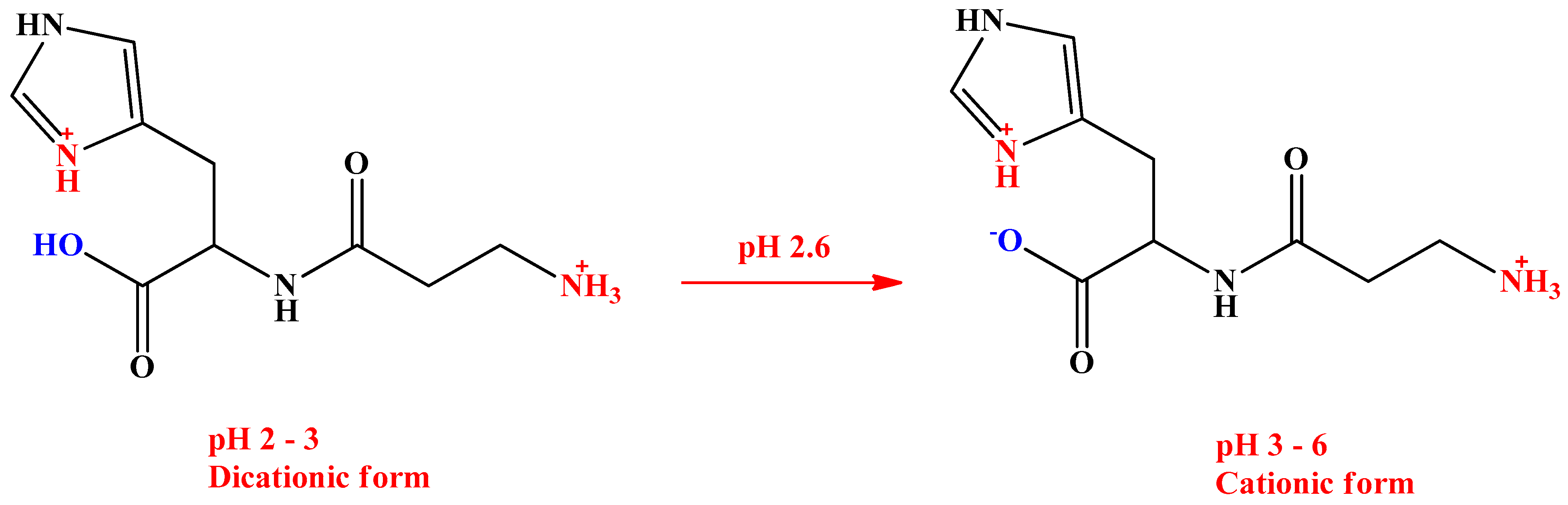
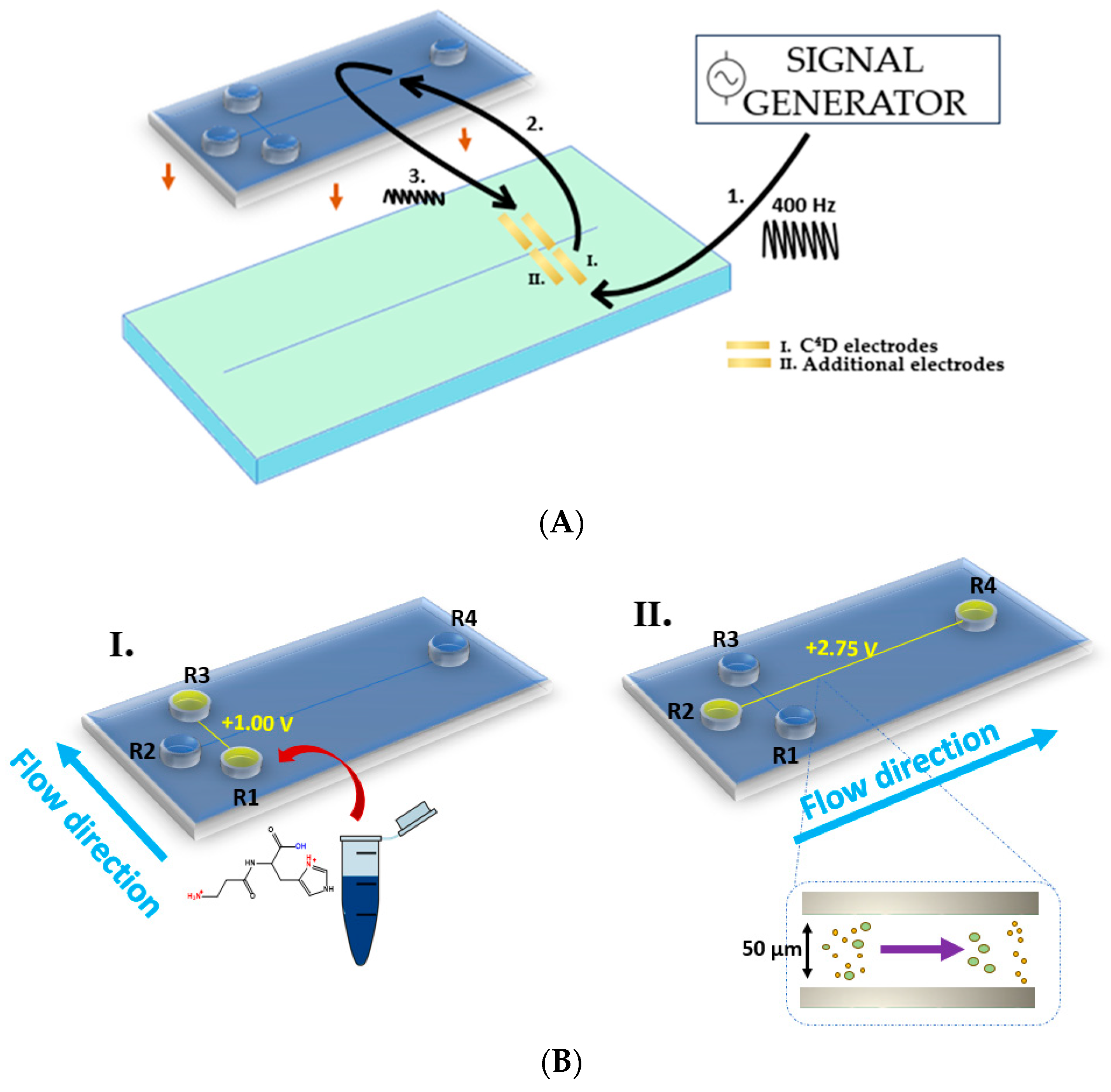
2.1. Method Optimization
2.2. Analytical Features of the Method
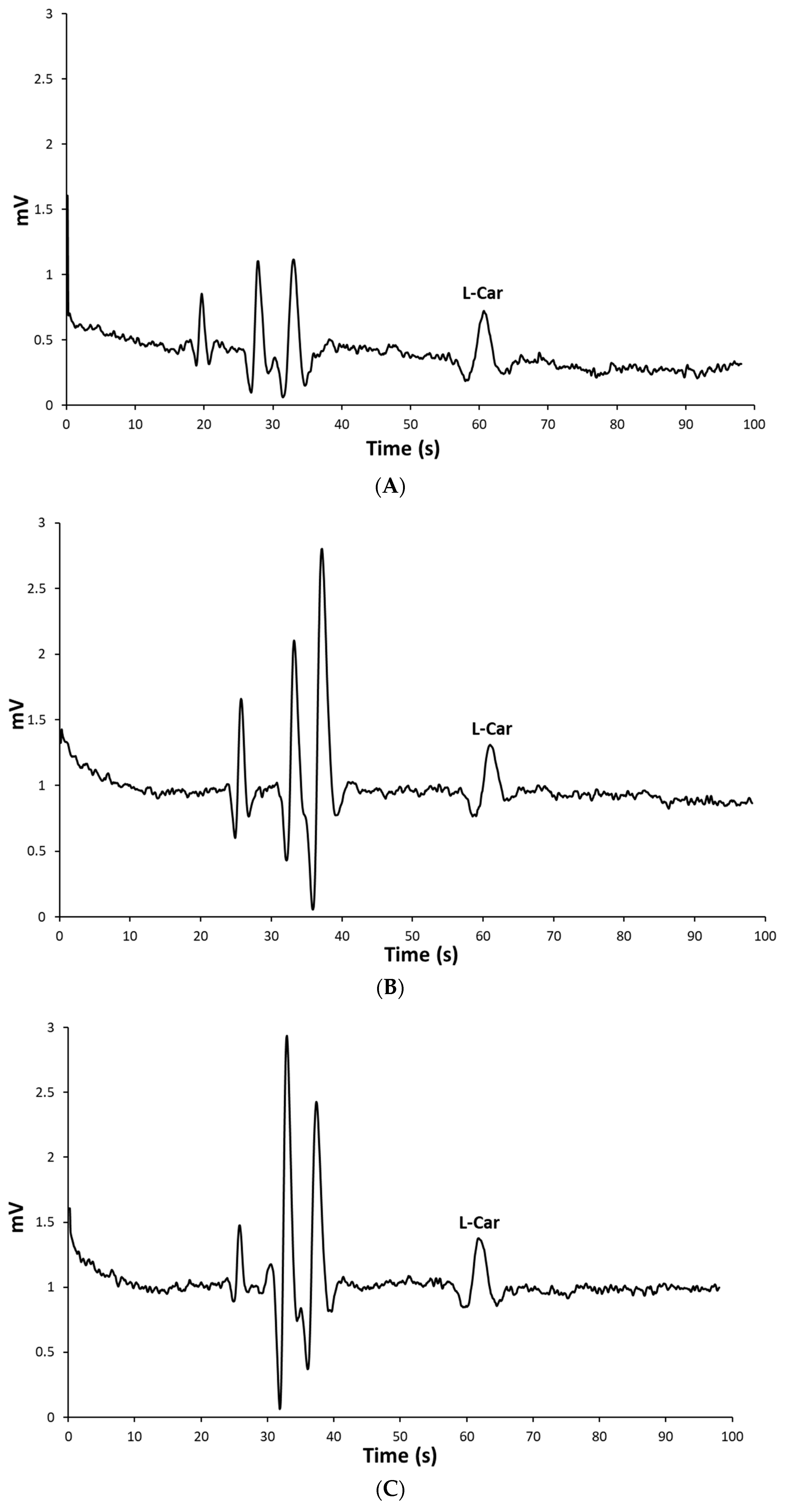
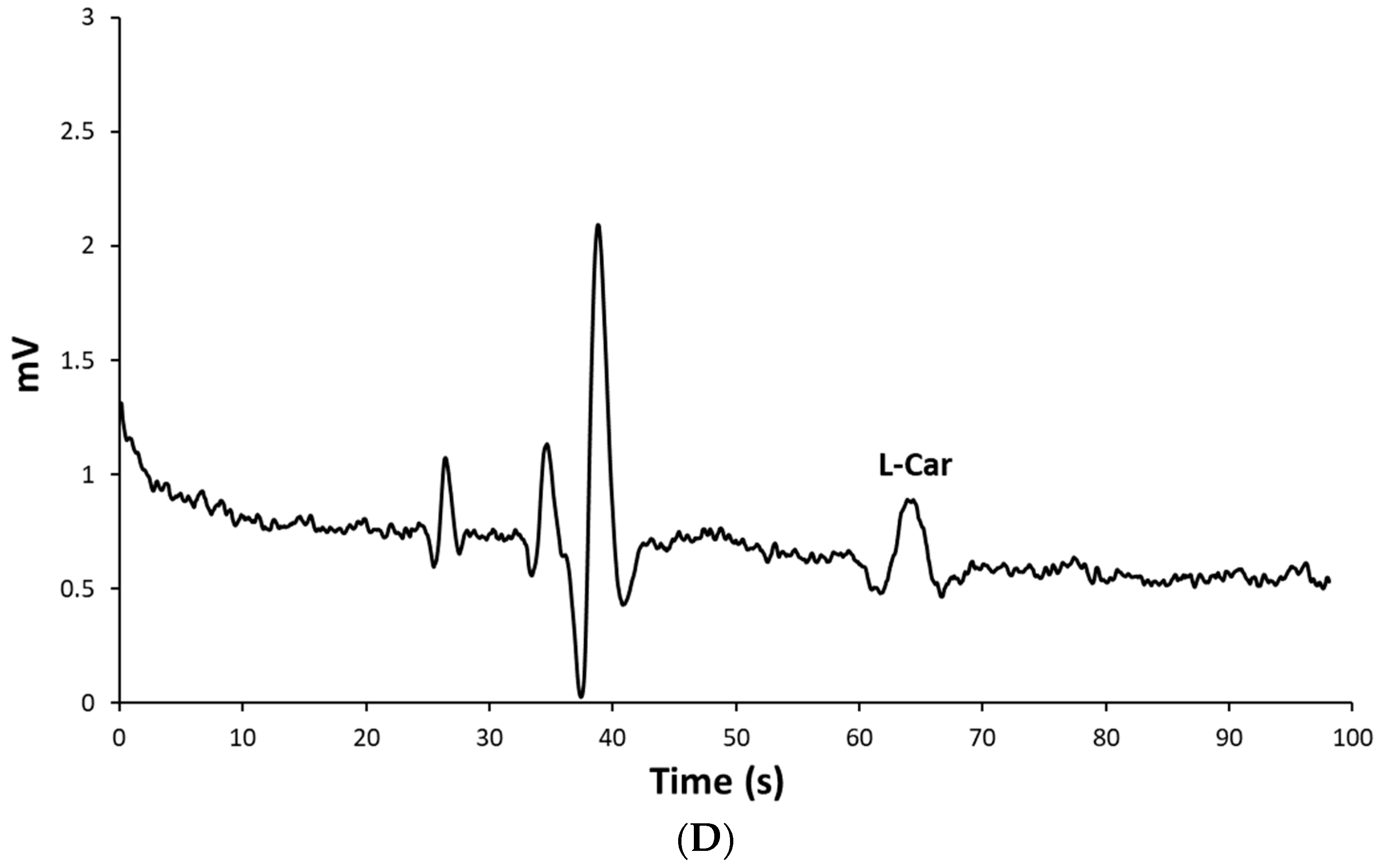
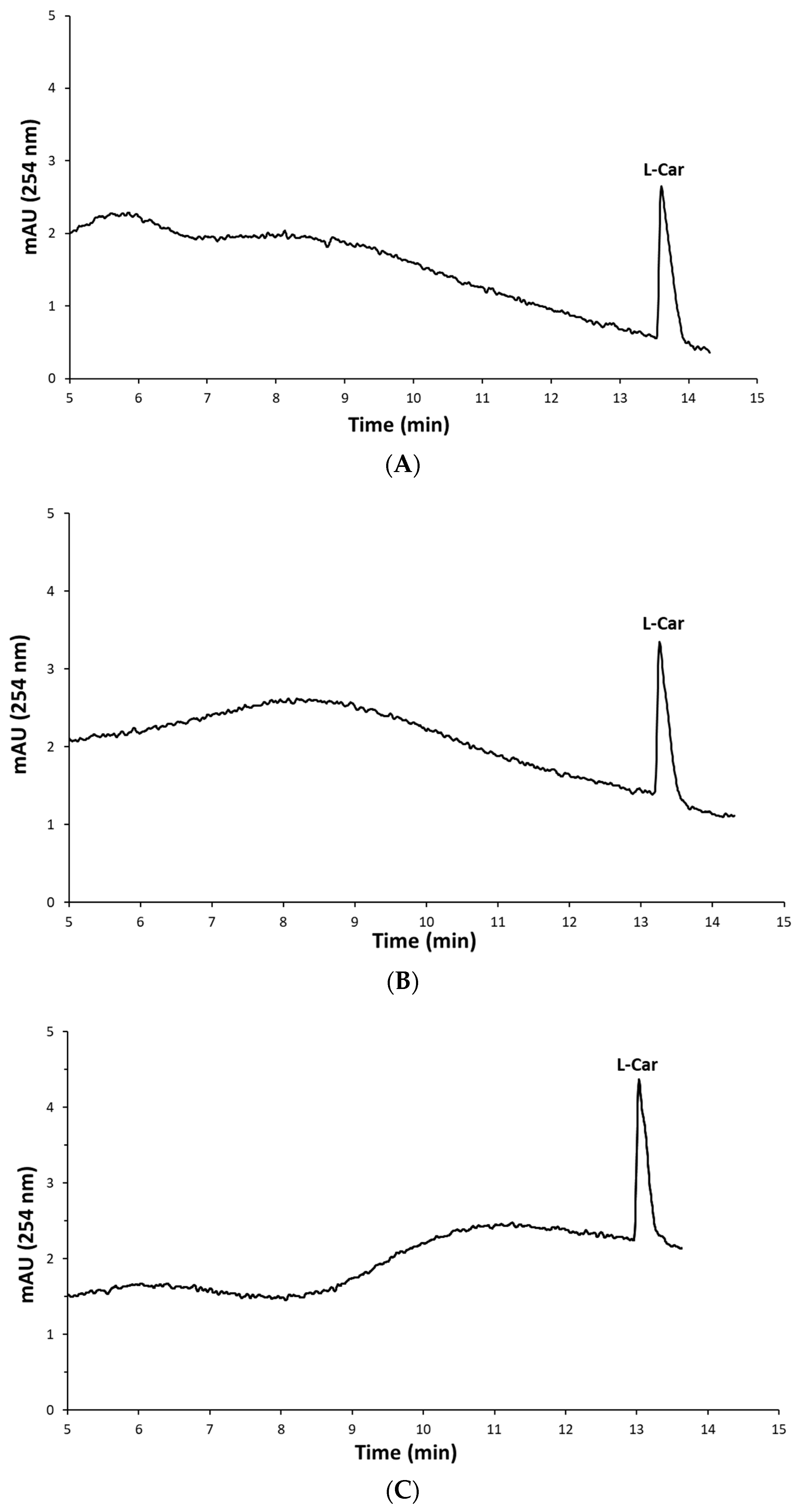
2.3. Determination of L-Carnosine in Real Samples
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Real Samples Preparation for MCE–C4D Analysis
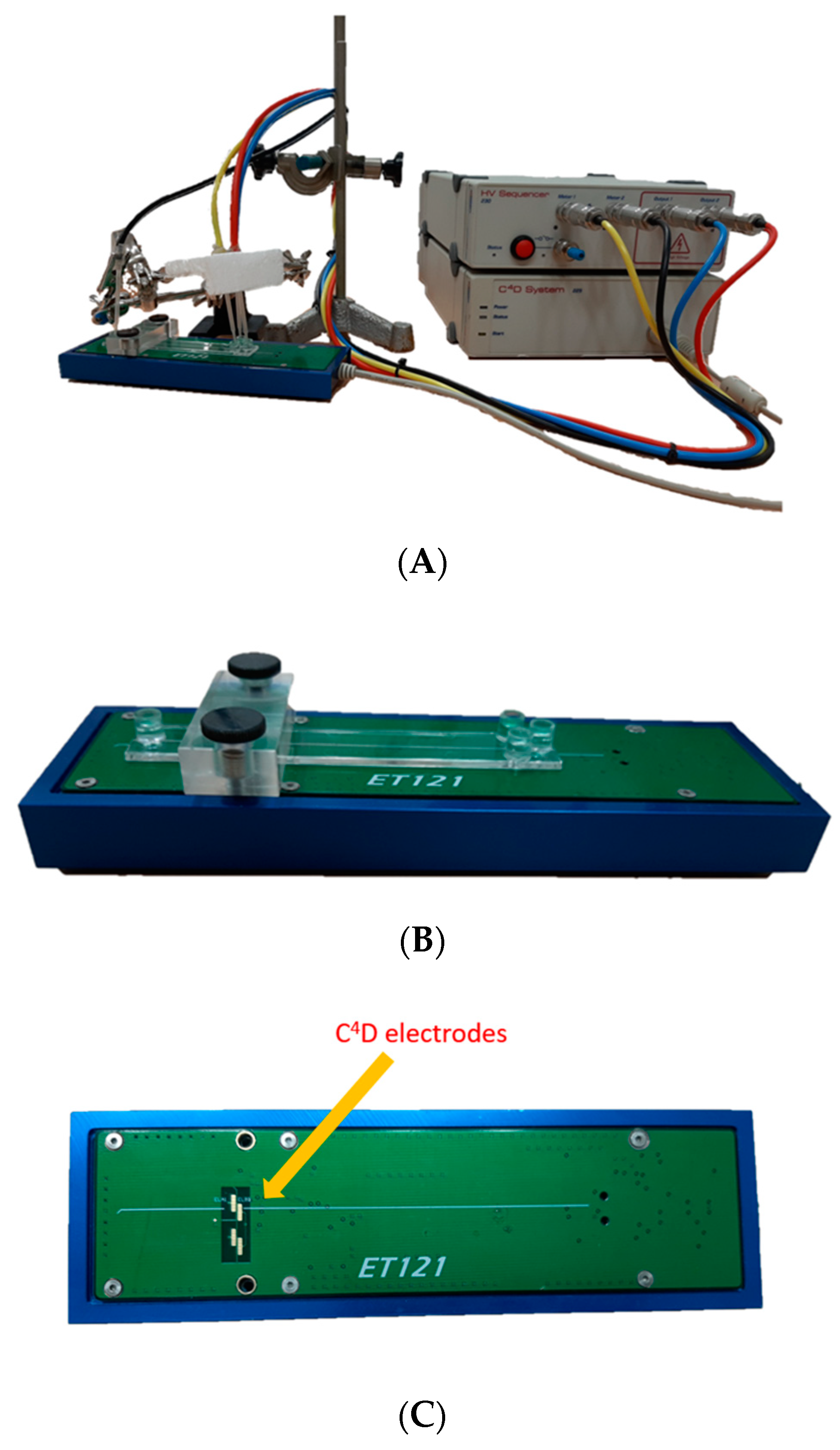
3.3. MCE–C4D Analysis
3.4. CE-UV-VIS Analysis
3.5. HPLC-DAD Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharma, R.; Sehrawat, R.; Ahlawat, S.; Sharma, V.; Parmar, A.; Thakur, M.S.; Mishra, A.K.; Tantia, M.S. An attempt to valorize the only black meat chicken breed of India by delineating superior functional attributes of its meat. Sci. Rep. 2022, 12, 3555. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.; Krapf, R.; Kuballa, T.; Bunzel, M. Differentiation of meat species of raw and processed meat based on polar metabolites using 1H NMR spectroscopy combined with multivariate data analysis. Front. Nutr. 2022, 30, 985797. [Google Scholar] [CrossRef] [PubMed]
- Aiello, G.; Rescigno, F.; Meloni, M.; Baron, G.; Aldini, G.; Carini, M.; D’Amato, A. Oxidative Stress Modulation by Carnosine in Scaffold Free Human Dermis Spheroids Model: A Proteomic Study. Int. J. Mol. Sci. 2022, 23, 1468. [Google Scholar] [CrossRef] [PubMed]
- Hasanein, P.; Felegari, Z. Chelating effects of carnosine in ameliorating nickel-induced nephrotoxicity in rats. Can. J. Physiol. Pharmacol. 2017, 95, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Kim, E.-S.; Kim, D.; Burrows, D.; De Felice, M.; Kim, M.; Baek, S.-H.; Ali, A.; Redgrave, J.; Doeppner, T.R.; et al. Comparative Cerebroprotective Potential of D- and L-Carnosine Following Ischemic Stroke in Mice. Int. J. Mol. Sci. 2020, 21, 3053. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, R.; Cameron, J.; Menon, K.; Mesinovic, J.; Jansons, P.; Scott, D.; Lu, Z.X.; de Courten, M.; Feehan, J.; de Courten, B.; et al. Carnosine Supplementation Improves Glucose Control in Adults with Pre-Diabetes and Type 2 Diabetes: A Randomised Controlled Trial. Available online: https://www.medrxiv.org/content/10.1101/2023.03.18.23287432v1 (accessed on 22 September 2023).
- Ansari, F.A.; Khan, A.A.; Mahmood, R. Ameliorative effect of carnosine and N-acetylcysteine against sodium nitrite induced nephrotoxicity in rats. J. Cell. Biochem. 2019, 120, 7032–7044. [Google Scholar] [CrossRef]
- Kawahara, M.; Sadakane, Y.; Mizuno, K.; Kato-Negishi, M.; Tanaka, K. Carnosine as a Possible Drug for Zinc-Induced Neurotoxicity and Vascular Dementia. Int. J. Mol. Sci. 2020, 21, 2570. [Google Scholar] [CrossRef]
- Hewlings, S.; Kalman, D. A review of zinc-L-carnosine and its positive effects on oral mucositis, taste disorders, and gastrointestinal disorders. Nutrients 2020, 12, 665. [Google Scholar] [CrossRef]
- Kim, S.K.; Kwon, D.; Kwon, D.A.; Paik, I.K.; Auh, J.H. Optimizing Carnosine Containing Extract Preparation from Chicken Breast for Anti-glycating agents. Korean J. Food Sci. 2014, 34, 127–132. [Google Scholar] [CrossRef]
- Cao, M.; Han, Q.; Zhang, J.; Zhang, R.; Wang, J.; Gu, W.; Kang, W.; Lian, K.; Ai, L. Meat differentiation between pasture-fed and concentrate-fed sheep/goats by liquid chromatography quadrupole time-of-flight mass spectrometry combined with metabolomic and lipidomic profiling. J. Chromatogr. A 2020, 1610, 460553. [Google Scholar] [CrossRef]
- Gatti, R.; Andreatta, P.; Boschetti, S. An high-performance liquid chromatographic method for the simultaneous analysis of acetylcarnitine taurinate, carnosine, asparagine and potassium aspartate and for the analysis of phosphoserine in alimentary supplements. J. Chromatogr. A 2013, 1298, 95–102. [Google Scholar] [CrossRef] [PubMed]
- do Nascimento Fontes, J.E.; Rodriguez-Caturla, M.Y.; Sant’Ana, A.S.; Lopes, T.I.B.; Marsaioli, A.J. Foodomics and storage monitoring of three meat cuts by 1H NMR. Concepts Magn. Reason. Part. A Bridg. Educ. Res. 2019, 47A, E21474. [Google Scholar] [CrossRef]
- Zikmundová, J.; Tůma, P.; Opekar, F. A dual spectrophotometric/contactless conductivity detector for CE determination of incompletely separated amino acids. J. Sep. Sci. 2008, 31, 353–355. [Google Scholar] [CrossRef]
- Duong, H.A.; Vu, M.T.; Nguyen, T.D.; Nguyen, M.H.; Mai, T.D. Determination of 10-hydroxy-2-decenoic acid and free amino acids in royal jelly supplements with purpose-made capillary electrophoresis coupled with contactless conductivity detection. J. Food Compost. Anal. 2020, 87, 103422. [Google Scholar] [CrossRef]
- Pormsila, W.; Krähenbühl, S.; Hauser, P.C. Determination of carnitine in food and food supplements by capillary electrophoresis with contactless conductivity detection. Electrophoresis 2010, 31, 2186–2191. [Google Scholar] [CrossRef]
- Staňová, A.; Marák, J.; Rezeli, M.; Páger, C.; Kilár, F.; Kaniansky, D. Analysis of therapeutic peptides in human urine by combination of capillary zone electrophoresis–electrospray mass spectrometry with preparative capillary isotachophoresis sample pretreatment. J. Chromatogr. A 2011, 1218, 8701–8707. [Google Scholar] [CrossRef]
- Nardiello, D.; Cataldi, T.R.I. Determination of carnosine in feed and meat by high-performance anion-exchange chromatography with integrated pulsed amperometric detection. J. Chromatogr. A 2004, 1035, 285–289. [Google Scholar] [CrossRef]
- Huang, Y.; Duan, J.; Chen, H.; Chen, M.; Chen, G. Separation and determination of carnosine-related peptides using capillary electrophoresis with laser-induced fluorescence detection. Electrophoresis 2005, 26, 593–599. [Google Scholar] [CrossRef]
- He, L.; Xu, Z.; Hirokawa, T.; Shen, L. Simultaneous determination of aliphatic, aromatic and heterocyclic biogenic amines without derivatization by capillary electrophoresis and application in beer analysis. J. Chromatogr. A 2017, 1482, 109–114. [Google Scholar] [CrossRef]
- Zhao, S.; Huang, Y.; Shi, M.; Huang, J.; Liu, Y.M. Quantification of carnosine-related peptides by microchip electrophoresis with chemiluminescence detection. Anal. Biochem. 2009, 393, 105–110. [Google Scholar] [CrossRef]
- Fresta, C.G.; Hogard, M.L.; Caruso, G.; Melo Costa, E.E.; Lazzarino, G.; Lunte, S.M. Monitoring carnosine uptake by RAW 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal. Methods 2017, 9, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xia, L.; Xiao, X.; Li, G. Enhanced capillary zone electrophoresis in cyclic olefin copolymer microchannels using the combination of dynamic and static coatings for rapid analysis of carnosine and niacinamide in cosmetics. J. Sep. Sci. 2022, 45, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Tůma, P.; Opekar, F.; Dlouhý, P. Capillary and microchip electrophoresis with contactless conductivity detection for analysis of foodstuffs and beverages. Food Chem. 2022, 375, 131858. [Google Scholar] [CrossRef] [PubMed]
- Tůma, P. Monitoring of biologically active substances in clinical samples by capillary and microchip electrophoresis with contactless conductivity detection: A review. Anal. Chim. Acta 2022, 1225, 340161. [Google Scholar] [CrossRef] [PubMed]
- Jozanović, M.; Hajduković, M.; Galović, O.; Kralik, G.; Kralik, Z.; Sakač, N.; Medvidović-Kosanović, M.; Sak-Bosnar, M. Determination of anti-oxidative histidine dipeptides in poultry by microchip capillary electrophoresis with contactless conductivity detection. Food Chem. 2017, 221, 1658–1665. [Google Scholar] [CrossRef]
- Jozanović, M.; Sakač, N.; Sak-Bosnar, M.; Carrilho, E. A simple and reliable new microchip electrophoresis method for fast measurements of imidazole dipeptides in meat from different animal species. Anal. Bioanal. Chem. 2018, 410, 4359–4369. [Google Scholar] [CrossRef]
- Domínguez-Vega, E.; Martínez-Girón, A.B.; Ruiz, C.G.; Crego, A.L.; Marina, M.L. Fast derivatization of the non-protein amino acid ornithine with FITC using an ultrasound probe prior to enantiomeric determination in food supplements by EKC. Electrophoresis 2009, 30, 1037–1045. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Z.; Lin, X.; Wen, Z.; Wang, F. Simultaneous analysis of seven oligopeptides in microbial fuel cell by micro-fluidic chip with reflux injection mode. Talanta 2012, 100, 338–343. [Google Scholar] [CrossRef]
- Cong, Y.; Katipamula, S.; Geng, T.; Prost, S.A.; Tang, K.; Kelly, R.T. Electrokinetic sample preconcentration and hydrodynamic sample injection for microchip electrophoresis using a pneumatic microvalve. Electrophoresis 2016, 37, 455–462. [Google Scholar] [CrossRef]
- Liang, R.P.; Wang, X.N.; Liu, C.M.; Meng, X.Y.; Qiu, J.D. Facile preparation of protein stationary phase based on polydopamine/graphene oxide platform for chip-based open tubular capillary electrochromatography enantioseparation. J. Chromatogr. A 2014, 1323, 135–142. [Google Scholar] [CrossRef]
- Liaoa, T.; Guoa, Z.; Lia, J.; Liua, M.; Chen, Y. One-step packing of anti-voltage photonic crystals into microfluidic channels for ultra-fast separation of amino acids and peptides. Lab. Chip 2013, 13, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Huang, H.; Zhao, J.; Li, S.; Liu, R.; Zhao, S. Quantification of glutathione in single cells from rat liver by microchip electrophoresis with chemiluminescence detection. Talanta 2018, 179, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Herzog, C.; Poehler, E.; Peretzki, A.J.; Borisov, S.M.; Aigner, D.; Mayr, T.; Nagl, S. Continuous on-chip fluorescence labelling, freeflow isoelectric focusing and marker-free isoelectric point determination of proteins and peptides. Lab. Chip 2016, 16, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Shimura, K.; Takahashi, K.; Koyama, Y.; Sato, K.; Kitamori, T. Isoelectric Focusing in a Microfluidically Defined Electrophoresis Channel. Anal. Chem. 2008, 80, 3818–3823. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, W.; Wang, W.; Xu, L.; Chen, G.; Fu, F. Separation and determination of β-casomorphins by using glass microfluidic chip electrophoresis together with laser-induced fluorescence detection. J. Sep. Sci. 2011, 34, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Nordman, N.; Lauren, S.; Kotiaho, T.; Franssila, S.; Kostiainen, R.; Sikanen, T. Interfacing microchip isoelectric focusing with on-chip electrospray ionization mass spectrometry. J. Chromatogr. A 2015, 1398, 121–126. [Google Scholar] [CrossRef]
- Wu, J.-K.; Yang, C.-S.; Wu, Y.-S.; Wang, P.-C.; Tseng, F.-G. Continuous affinity-gradient nano-stationary phase served as a column for reversed-phase electrochromatography and matrix carrier in time-of-flight mass spectrometry for protein analysis. Anal. Chim. Acta 2015, 889, 166–171. [Google Scholar] [CrossRef]
- Chong, K.C.; Thang, L.Y.; Quirino, J.P.; See, H.H. Monitoring of vancomycin in human plasma via portable microchip electrophoresis with contactless conductivity detector and multi-stacking strategy. J. Chromatogr. A 2017, 1485, 142–146. [Google Scholar] [CrossRef]
- Reschke, B.R.; Schiffbauer, J.; Edwardsb, B.F.; Timperman, A.T. Simultaneous separation and detection of cations and anions on a microfluidic device with suppressed electroosmotic flow and a single injection point. Analyst 2010, 135, 1351–1359. [Google Scholar] [CrossRef]
- Yamamoto, S.; Himeno, M.; Kobayashi, M.; Akamatsu, M.; Satoh, R.; Kinoshita, M.; Sugiura, R.; Suzuki, S. Microchip electrophoresis utilizing an in situ photopolymerized Phos-tag binding polyacrylamide gel for specific entrapment and analysis of phosphorylated compounds. Analyst 2017, 142, 3416–3423. [Google Scholar] [CrossRef]
- Araya-Farias†, M.; Dziomba†, S.; Carbonnier, B.; Guerrouache, M.; Ayed, I.; Aboud, N.; Taverna, M.; Tran, N.T. A lab-on-a-chip for monolith-based preconcentration and electrophoresis separation of phosphopeptides. Analyst 2017, 142, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, E.; Bonabi, A.; Nordman, N.; Jokinen, V.; Kotiaho, T.; Kostiainen, R.; Sikanen, T. Rapid separation of phos-phopeptides by microchip electrophoresis–electrospray ionization mass spectrometry. J. Chromatogr. A 2016, 1440, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Xia, Z.; Wei, W.; Gou, Q. Direct UV detection of underivatized amino acids using capillary electrophoresis with online sweeping enrichment. J. Sep. Sci. 2009, 32, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Steed, R. Manual of Analysis of Amino Acids by HPLC; Agilent Technologies, Inc.: Santa Clara, CA, USA, 2010. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | MCE-C4D |
|---|---|
| Migration time (s) | 61 ± 0.42 |
| R2 | 0.9976 |
| Linear range (M) | 5 × 10−6 – 5 × 10−5 |
| Slope | 27,567 |
| Intercept * | 0.1852 |
| LOD (μM) | 2.5 × 10−6 |
| LOQ (μM) | 5 × 10−6 |
| Sample 1 | Sample 2 | Sample 3 | ||
|---|---|---|---|---|
| Declared Value (mg/g Sample) | 171.42 | 172.50 | 833.33 | |
| MCE-C4D analysis | Results (mg/g sample) | 184.53 ± 8.28 | 182.91 ± 15.95 | 965.79 ± 130.50 |
| RSD (%) | 4.49 | 8.72 | 13.51 | |
| % Deviation from the declared value | +7.65 ± 4.83 | +6.03 ± 9.24 | +15.95 ± 15.66 | |
| CE-UV-VIS analysis | Results (mg/g sample) | 178.60 ± 6.39 | 167.7 ± 4.57 | 927.72 ± 3.45 |
| RSD (%) | 3.58 | 2.72 | 0.37 | |
| % Deviation from the declared value | +4.19 ± 3.73 | −2.78 ± 2.65 | +11.33 ± 0.42 | |
| HPLC-DAD analysis | Results (mg/g sample) | 198.01 ± 9.80 | 177.4 ± 8.48 | 980.63 ± 47.10 |
| RSD (%) | 5 | 5 | 5 | |
| % Deviation from the declared value | +15.51 ± 5.71 | +2.84 ± 4.92 | +17.68 ± 5.65 |
| Unspiked Sample (μM) | Added Amount (μM) | Spiked Sample (μM) | Recovery (%) | |
|---|---|---|---|---|
| * Recovery experiment 1 | ||||
| Sample 1 | 11.01 ± 0.04 | 2.20 | 13.09 ± 0.07 | 96.66 ± 1.90 |
| Sample 2 | 10.19 ± 0.10 | 2.18 | 13.09 ± 0.07 | 99.73 ± 3.32 |
| Sample 3 | 8.53 ± 0.010 | 1.70 | 10.28 ± 0.08 | 102.36 ± 4.91 |
| * Recovery experiment 2 | ||||
| Sample 1 | 11.01 ± 0.04 | 4.40 | 15.76 ± 0.20 | 108.00 ± 4.54 |
| Sample 2 | 10.19 ± 0.10 | 4.36 | 15.22 ± 0.23 | 98.62 ± 5.29 |
| Sample 3 | 8.53 ± 0.010 | 3.41 | 12.12 ± 0.25 | 108.73 ± 3.20 |
| Characteristics | ME-C4D | CE-UV-VIS | HPLC-DAD |
|---|---|---|---|
| R2 | 0.9976 | 0.9905 | 0.9996 |
| Measured concentration range | 5 × 10−6–5 × 10−5 | 4.4 × 10−4–4.4 × 10−3 | 5 × 10−6–2.5 × 10−4 |
| Slope | 27,567 | 64.022 | 3.72 × 10−7 |
| Intercept * | 0.1852 | −3.938 | 2.43 × 10−6 |
| Analyte/Sample | BGE | Chip Material | Linear Range/Tested Range | LOD (Analyzed Conc.) (μM) | Detector | Ref. |
|---|---|---|---|---|---|---|
| βA-H (carnosine)/health supplements | 0.5 M HAc | PMMA | 5–50 | 2.5 | C4D | This work |
| βA-H (carnosine), homocarnosine (GABA-H) anserine (βA-methyl-H)/human cerebrospinal fluid and canine plasma | 15 mM borate buffer (pH 9.8) + 1.0 mM Co2+ + 1.0 mM adenine +35 mM SDS | glass/PDMS | 0.06–15 | 0.030 0.028 0.034 | CL | [21] |
| βA-H (carnosine) βA-methyl-H (anserine)/breast muscle | 200 mM HAc + 10 mM HIBA + 0.3 mM KOAc (pH 2.7) | PMMA | 0–200 | 0.10 0.16 | C4D | [26] |
| βA-H (carnosine)/macrophage cells | 20 mM borate (pH 9.2) | glass | 0.025–5 | 0.065 | LIF | [22] |
| βA-H (carnosine), βA-methyl-H (anserine)/meat | 250 Mm HAc + 10 mM 2-OH-n-butyric acid + 0.3 mM RbOAc (pH 2.6) | PMMA | 0–88.42 | 0.038 0.069 | C4D | [27] |
| βA-H (carnosine)/cosmetics | 0.1 mM borax with + 0.03% w/w SDS | BSA- COC | 0.38–226.24 mg/kg | 0.17 mg/kg, (0.10, 10) | LIF | [23] |
| GG, GL, RPP, KPV, VKK WYD, YWS/ anode chamber of microbial fuel | 10 mM borax-H3BO3 buffer + 20 mM SDS (pH = 9.0) | ITO-glass | 0.625–10 0.625–10 0.31–10 1.25–20 1.25–20 0.625–10 0.625–10 | 0.050 0.050 0.025 0.625 0.100 0.050 0.050 | LIF | [29] |
| GF isomers/ model solution | 20 mM PBS (pH 7.17) | PDA/ GO/BSA-PDMS | N/A | N/A (4000) | Amperometry | [31] |
| FA, YGGFL (leucine enkephalin), GGYR/model solution | 10 mM Na2CO3 + 0.5% HPC (w/v) (pH 9.7) | HPC-PDMS | N/A 1–10 N/A | N/A (0.02) | Fluorescence | [30] |
| GGGG GGGGG GGGGGG/model solution | 20 mM Na2B4O7 (pH 10.02) | glass | N/A | N/A (10) | Fluorescence | [32] |
| GCE (glutathione)/ rat liver cells | 20 mM Na3PO4 + 1.5 mM luminol (pH 9.6) | glass | 3–600 | 0.96 | CL | [33] |
| XLYENKPRRPYIL (neurote-nsin), CYIQNCPLG (oxytocin), YGGFL (leucine enkephalin), EADPNK + FYGLM (tryptic digestion of physalaemin)/model solution | Ampholyte mixture (pH 4–7) | acrylate-glass | N/A | N/A (100, 100, 100, 966) | Fluorescence | [34] |
| pI markers: GCDDD (pI 3.64), GCHQHQHQHQ (pI 53) * GCQHHHR (pI 7.58) * GCYYYKK (pI 9.56)/model solution | 2.5% (v/v) Pharmalyte 3–10 | glass | N/A | 3 × 10−6 for the pI 5.53 (0.0025) | LIF | [35] |
| β-casomorphins: YPFPG, TPFPGP, YPFPGPI, TPFPG, PFPGPI/cheese | 15 mM H3BO3- Na2H20B4O17 (pH 10.5) | glass | 0.01–1 0.05–1 0.05–1 0.03–1 0.05–1 | 0.075 0.042 0.024 0.019 0.019 | LIF | [36] |
| DRVYIHPFHL (Angiotensin I), DRVYIHPF (Angiotensin II), RVYIHPF (Angiotensin III), RPPGF (Bradykinin fragment 1–5)/model solution | 30 mM NH4Ac + 30% MeOH | PDMS | N/A | N/A (100) | ESI/MS | [37] |
| RPPGFSP (Bradykinin fragment 1–7), SYSMEHFRW, GKPVGKKR (ACTH fragment 1–17)/model solution | ACN: 10 mmol/L Tris buffer (pH 8.2)/80:20 | NPs -silicon | N/A | N/A (6) | MALDI-TOF-MS | [38] |
| Vancomycin (7 modified AAs); (NLEU)-(HDPY)-(ASN)–p-(DPYG)-(DHPG)-HDPY-(LPGH)/human plasma | 300 mM HAc | glass | 5.18–4.86 | 0.83 | C4D | [39] |
| Ac-R-[CMLNRVYRPC]/model solution | 25 mM phosphate buffer (pH 7) | PEG- glass | N/A | N/A (100) | Fluorescence | [40] |
| PO4-glycogen synthase (PLSRTLSVSS)/model solution | 100 mM Tris phosphate buffer (pH 7.0)/10 mM EDTA | PMMA | 0.001–0.025 | 0.001 | Fluorescence | [41] |
| PO4-peptides: DHTGFLpTEYVATR, DHTGFLTEpYVATR, DHTGFLpTEpYVATR, Non-PO4-peptide: DHTGFLTEYVATR | 25 mM borax + 2.8% NH4OH (pH 11.5) | glass | 0.04–1.34 | 0.015 for PO4-peptides (0.31) | Fluorescence | [42] |
| Insulin receptor peptide: Non-PO4-peptides: TRDIYETDYYRK PO4-peptides: TRDIpYETDYYRK TRDIYETDpYYRK TRDIpYETDpYpYRK | 40 H2O/60 MeOH (v/v) + 30 mM NH4-Ac (pH 7.4) | PDMS | N/A | N/A (125, 250) | ESI-MS | [43] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pukleš, I.; Páger, C.; Sakač, N.; Šarkanj, B.; Matasović, B.; Samardžić, M.; Budetić, M.; Marković, D.; Jozanović, M. Electrophoretic Determination of L-Carnosine in Health Supplements Using an Integrated Lab-on-a-Chip Platform with Contactless Conductivity Detection. Int. J. Mol. Sci. 2023, 24, 14705. https://doi.org/10.3390/ijms241914705
Pukleš I, Páger C, Sakač N, Šarkanj B, Matasović B, Samardžić M, Budetić M, Marković D, Jozanović M. Electrophoretic Determination of L-Carnosine in Health Supplements Using an Integrated Lab-on-a-Chip Platform with Contactless Conductivity Detection. International Journal of Molecular Sciences. 2023; 24(19):14705. https://doi.org/10.3390/ijms241914705
Chicago/Turabian StylePukleš, Iva, Csilla Páger, Nikola Sakač, Bojan Šarkanj, Brunislav Matasović, Mirela Samardžić, Mateja Budetić, Dean Marković, and Marija Jozanović. 2023. "Electrophoretic Determination of L-Carnosine in Health Supplements Using an Integrated Lab-on-a-Chip Platform with Contactless Conductivity Detection" International Journal of Molecular Sciences 24, no. 19: 14705. https://doi.org/10.3390/ijms241914705
APA StylePukleš, I., Páger, C., Sakač, N., Šarkanj, B., Matasović, B., Samardžić, M., Budetić, M., Marković, D., & Jozanović, M. (2023). Electrophoretic Determination of L-Carnosine in Health Supplements Using an Integrated Lab-on-a-Chip Platform with Contactless Conductivity Detection. International Journal of Molecular Sciences, 24(19), 14705. https://doi.org/10.3390/ijms241914705