
Influence of Race and High Laminar Shear Stress on TNFR1 Signaling in Endothelial Cells
 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
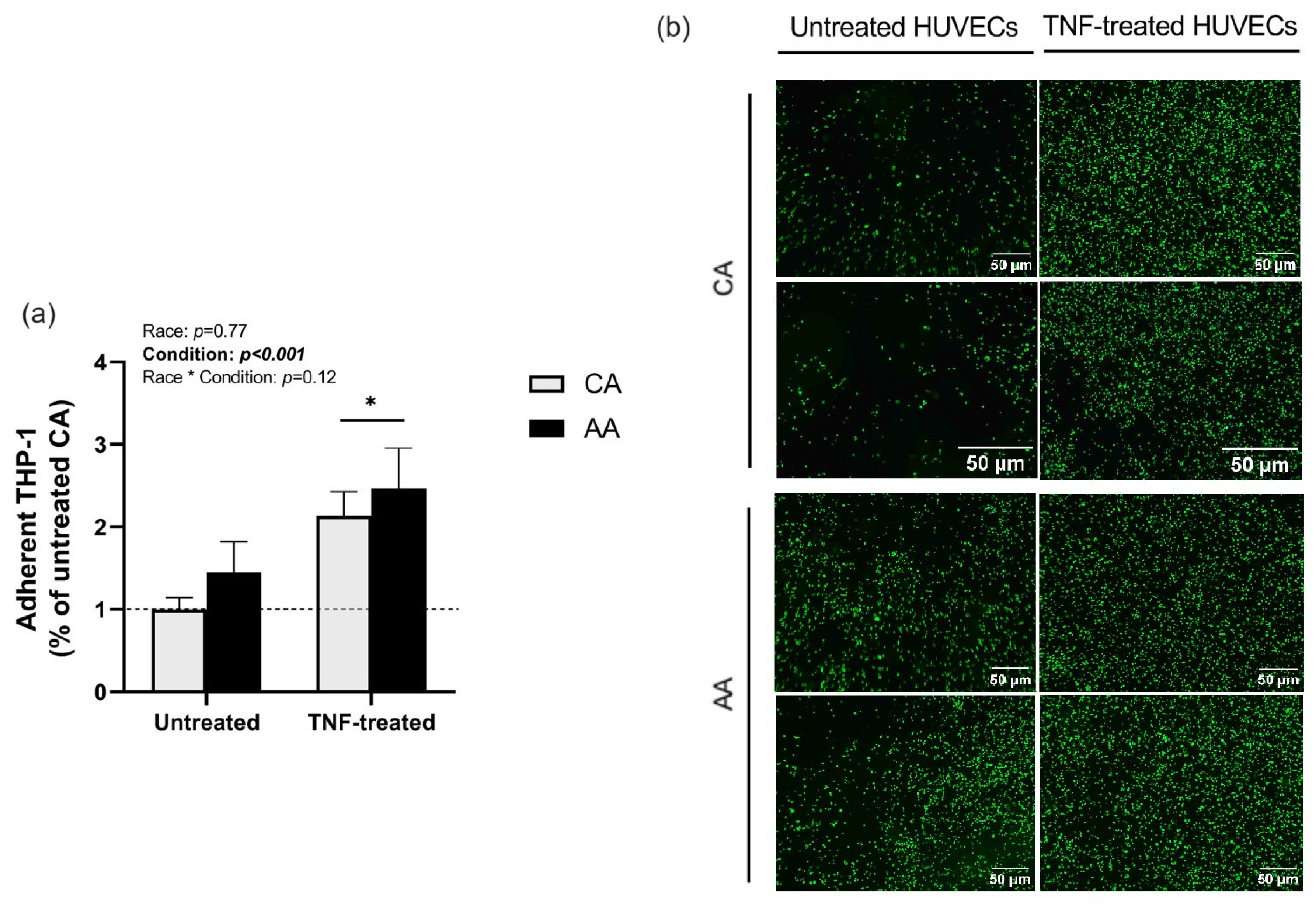
2.1. THP-1 Monocyte Adhesion
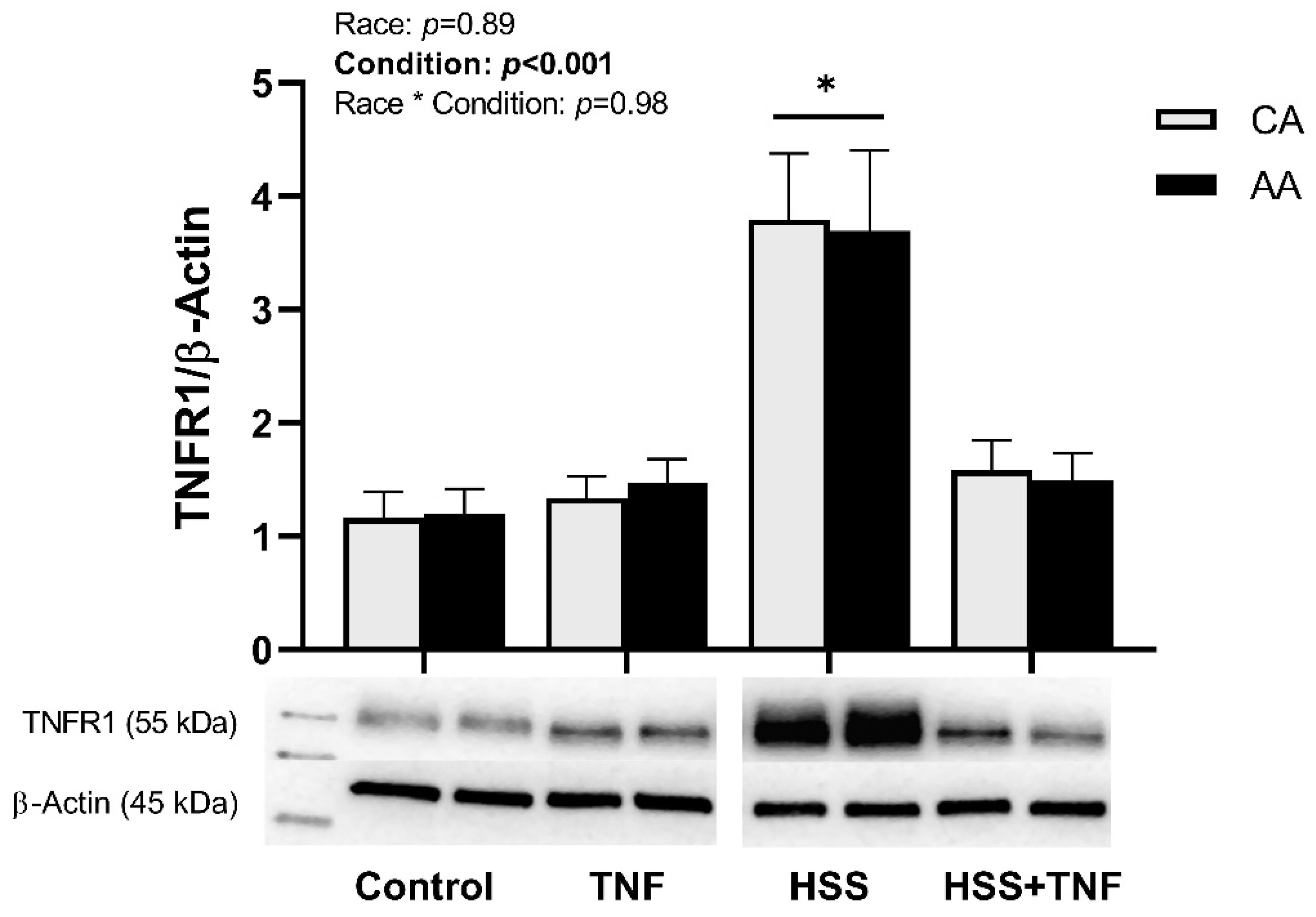
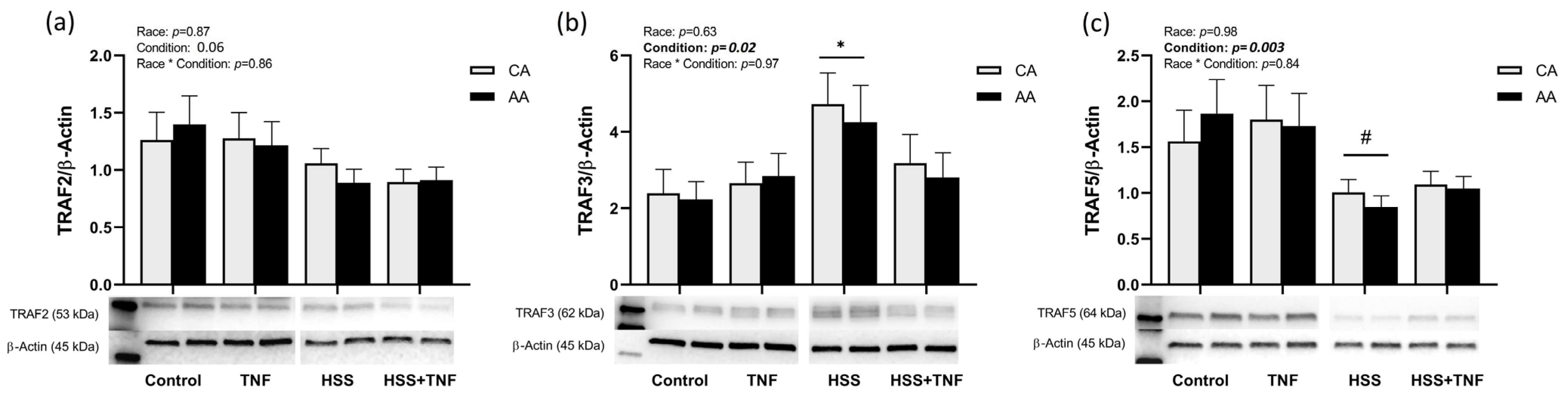
2.2. TNFR1 Signaling Complex and HSS
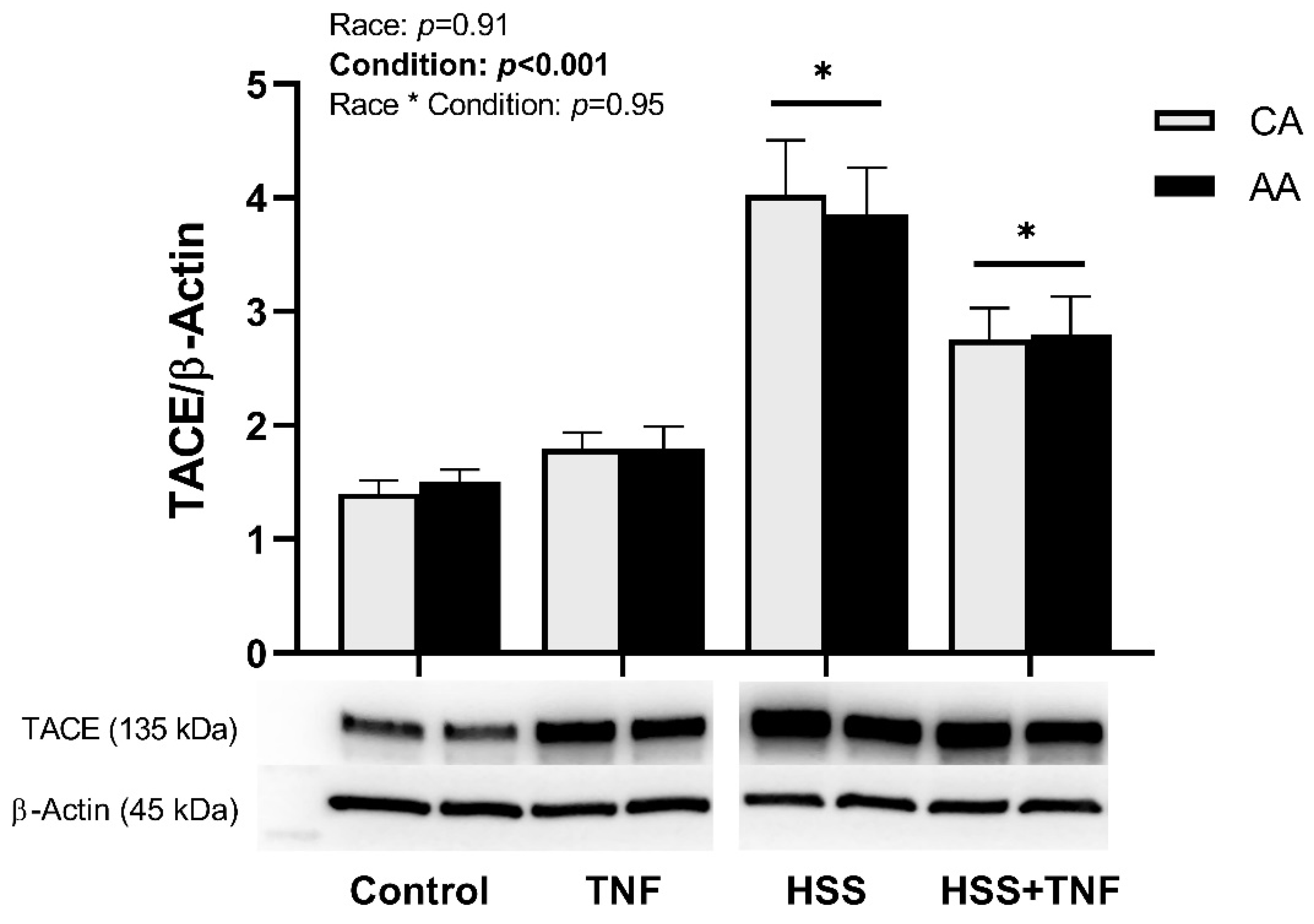
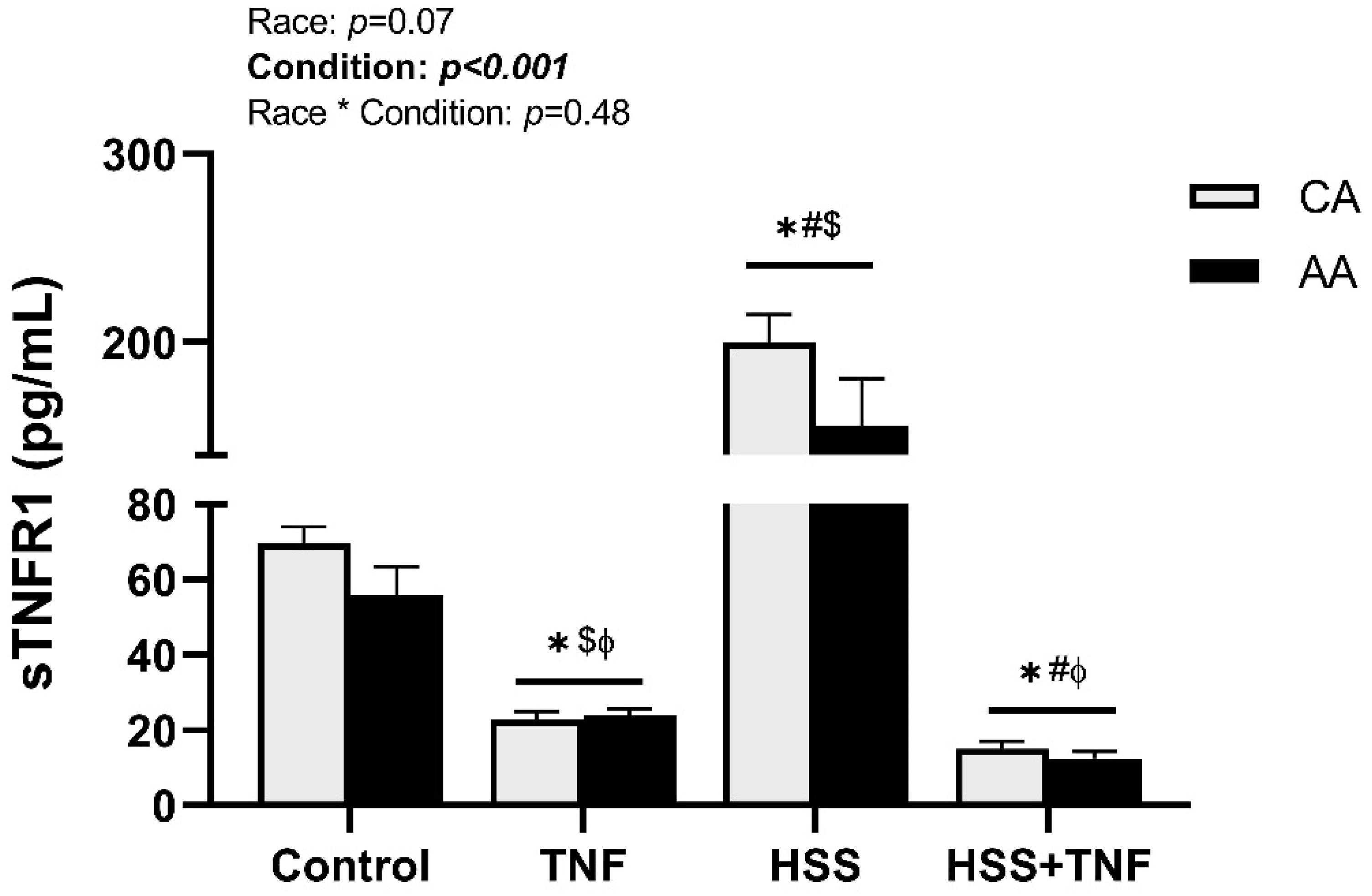
2.3. TNFR1 Shedding and HSS
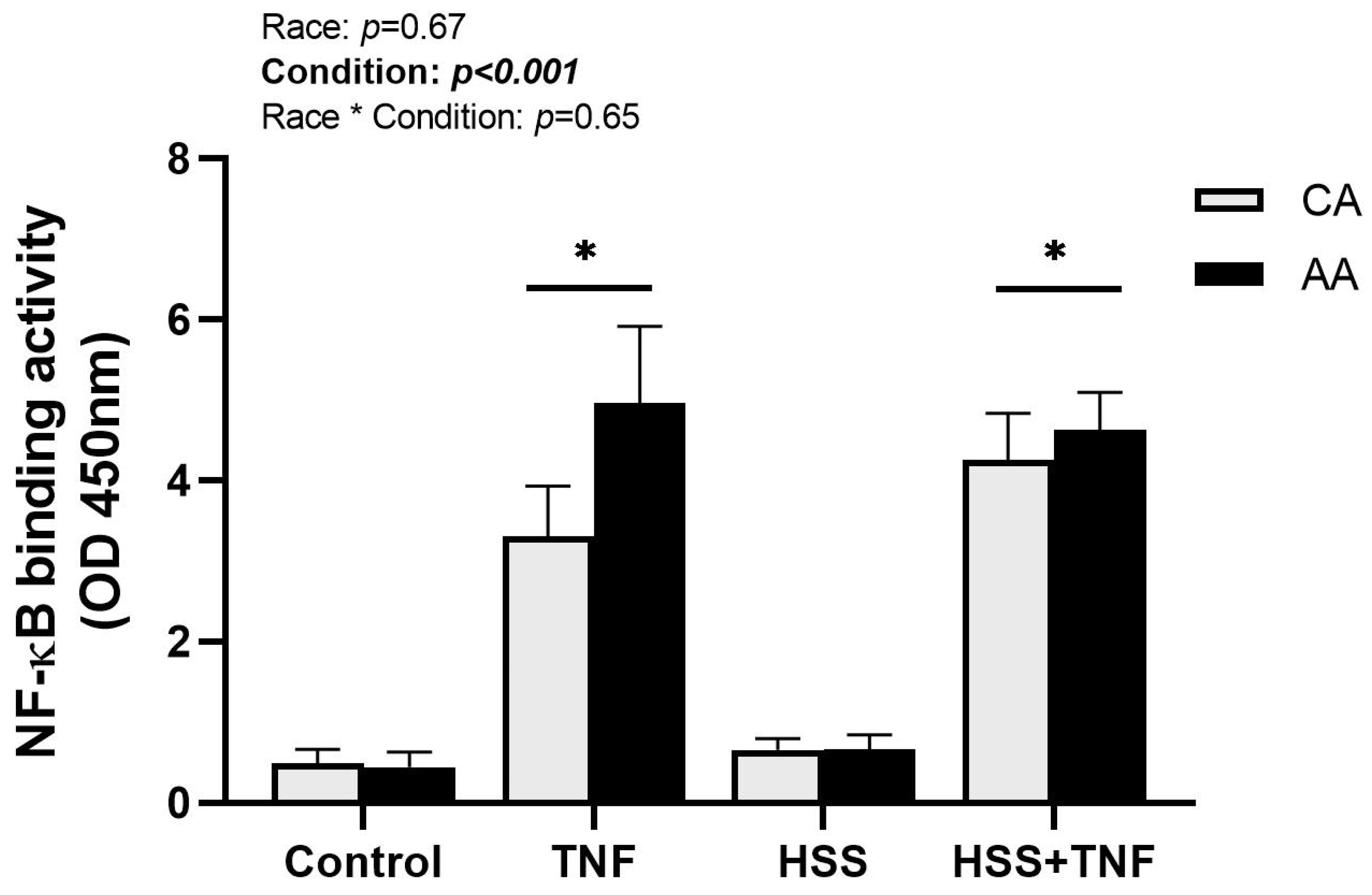
2.4. NF-κB Binding Activity
3. Discussion
Limitations
4. Materials and Methods
4.1. Cell Culture
4.1.1. HUVECs
4.1.2. THP-1
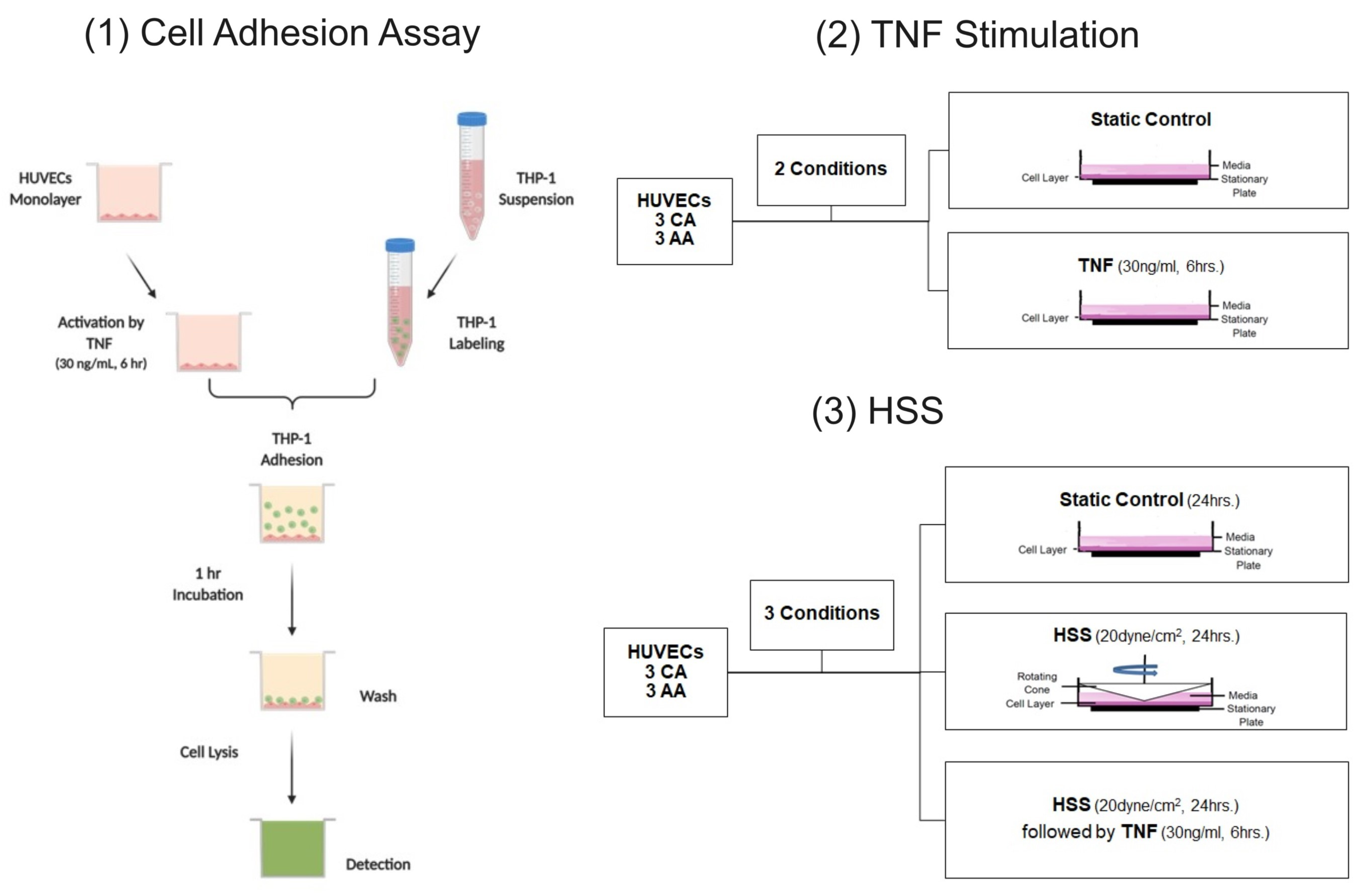
4.2. Cell Adhesion Assay
4.3. Laminar Shear Stress
4.4. Western Blotting
4.5. Assays
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| TNF | Tumor necrosis factor |
| TNFR I | TNF receptor I |
| TRAF | TNFR-associated factor |
| NF-κB | Nuclear factor of kappa B |
| MAP Kinase | Mitogen-activated protein kinase |
| JNK | c-Jun N-terminal kinases |
| eNOS | Endothelial nitric oxide synthase |
| NO | Nitric oxide |
| sTNFR | Soluble TNF receptor |
| HSS | High laminar shear stress |
| AA | African American |
| ECs | Endothelial cells |
| CA | Caucasian American |
| CVD | Cardiovascular diseases |
| cIMT | Carotid intima-media thickness |
| FMD | Flow-mediated dilation |
| ET-1 | Endothelin-1 |
| HUVECs | Human umbilical vein endothelial cells |
| SOD1 | Superoxide dismutase 1 |
| IL-6 | Interleukin-6 |
| TACE | TNFα-converting enzyme |
| NIK | NF-κB-inducing kinase |
| cIAP1/2 | Cellular inhibitor of apoptosis protein 1/2 |
| AP-1 | Activator protein 1 |
| IL-1 | Interleukin-1 |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| ICAM-1 | Intracellular adhesion molecule 1 |
| MCP-1 | Monocyte chemoattractant protein 1 |
| PBMCs | Peripheral blood mononuclear cells |
References
- Madge, L.A.; Pober, J.S. TNF signaling in vascular endothelial cells. Exp. Mol. Pathol. 2001, 70, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kofler, S.; Nickel, T.; Weis, M. Role of cytokines in cardiovascular diseases: A focus on endothelial responses to inflammation. Clin. Sci. 2005, 108, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S. TNF as an activator of vascular endothelium. Ann. Inst. Pasteur Immunol. 1988, 139, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, M.; Rasool, H.F.; Javed, I.; Raza, S.; Abang, L.; Hashim, M.M.A.; Saleem, Z.; Abdullah, R.M.; Faraz, M.A.; Hassan, K.M.; et al. Comparative Roles of IL-1, IL-6, IL-10, IL-17, IL-18, 1L-22, IL-33, and IL-37 in Various Cardiovascular Diseases with Potential Insights for Targeted Immunotherapy. Cureus 2023, 15, e42494. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef]
- Walczak, H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol. Rev. 2011, 244, 9–28. [Google Scholar] [CrossRef]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef]
- Murdaca, G.; Spanò, F.; Cagnati, P.; Puppo, F. Free radicals and endothelial dysfunction: Potential positive effects of TNF-α inhibitors. Redox Rep. 2013, 18, 95–99. [Google Scholar] [CrossRef]
- Steyers, C.M., 3rd; Miller, F.J., Jr. Endothelial dysfunction in chronic inflammatory diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [PubMed]
- McKellar, G.E.; McCarey, D.W.; Sattar, N.; McInnes, I.B. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat. Rev. Cardiol. 2009, 6, 410–417. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef] [PubMed]
- Zirlik, A.; Bavendiek, U.; Libby, P.; MacFarlane, L.; Gerdes, N.; Jagielska, J.; Ernst, S.; Aikawa, M.; Nakano, H.; Tsitsikov, E.; et al. TRAF-1, -2, -3, -5, and -6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of CD40L in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Gertzberg, N.; Johnson, A. TNF-alpha induces a decrease in eNOS promoter activity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L452–L459. [Google Scholar] [CrossRef]
- Clarkson, P.; Montgomery, H.E.; Mullen, M.J.; Donald, A.E.; Powe, A.J.; Bull, T.; Jubb, M.; World, M.; Deanfield, J.E. Exercise training enhances endothelial function in young men. J. Am. Coll. Cardiol. 1999, 33, 1379–1385. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. The effects of exercise training on markers of endothelial function in young healthy men. Int. J. Sports Med. 2003, 24, 404–409. [Google Scholar] [CrossRef]
- Green, D.J.; Cable, N.T.; Fox, C.; Rankin, J.M.; Taylor, R.R. Modification of forearm resistance vessels by exercise training in young men. J. Appl. Physiol. 1994, 77, 1829–1833. [Google Scholar] [CrossRef]
- Fernandez-Real, J.M.; Lainez, B.; Vendrell, J.; Rigla, M.; Castro, A.; Peñarroja, G.; Broch, M.; Pérez, A.; Richart, C.; Engel, P.; et al. Shedding of TNF-alpha receptors, blood pressure, and insulin sensitivity in type 2 diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E952–E959. [Google Scholar] [CrossRef]
- Gomes, W.F.; Lacerda, A.C.; Mendonça, V.A.; Arrieiro, A.N.; Fonseca, S.F.; Amorim, M.R.; Rocha-Vieira, E.; Teixeira, A.L.; Teixeira, M.M.; Miranda, A.S.; et al. Effect of aerobic training on plasma cytokines and soluble receptors in elderly women with knee osteoarthritis, in response to acute exercise. Clin. Rheumatol. 2012, 31, 759–766. [Google Scholar] [CrossRef]
- Fonseca, T.R.; Mendes, T.T.; Ramos, G.P.; Cabido, C.E.T.; Morandi, R.F.; Ferraz, F.O.; Miranda, A.S.; Mendonça, V.A.; Teixeira, A.L.; Silami-Garcia, E.; et al. Aerobic Training Modulates the Increase in Plasma Concentrations of Cytokines in response to a Session of Exercise. J. Environ. Public Health 2021, 2021, 1304139. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R. The role of TNF in cardiovascular disease. Pharmacol. Res. 1999, 40, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, M.; Abe, J.; Tsuchiya, K.; Berk, B.C.; Tamaki, T. Stress and vascular responses: Atheroprotective effect of laminar fluid shear stress in endothelial cells: Possible role of mitogen-activated protein kinases. J. Pharmacol. Sci. 2003, 91, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Abe, J.I.; Min, W.; Surapisitchat, J.; Yan, C. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann. N. Y. Acad. Sci. 2001, 947, 93–109; discussion 109–111. [Google Scholar] [CrossRef]
- Yamawaki, H.; Lehoux, S.; Berk, B.C. Chronic physiological shear stress inhibits tumor necrosis factor-induced proinflammatory responses in rabbit aorta perfused ex vivo. Circulation 2003, 108, 1619–1625. [Google Scholar] [CrossRef]
- Brown, M.D.; Feairheller, D.L. Are there race-dependent endothelial cell responses to exercise? Exerc. Sport Sci. Rev. 2013, 41, 44–54. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Patel, P.D.; Velazquez, J.L.; Arora, R.R. Endothelial dysfunction in African-Americans. Int. J. Cardiol. 2009, 132, 157–172. [Google Scholar] [CrossRef]
- Campia, U.; Choucair, W.K.; Bryant, M.B.; Waclawiw, M.A.; Cardillo, C.; Panza, J.A. Reduced endothelium-dependent and -independent dilation of conductance arteries in African Americans. J. Am. Coll. Cardiol. 2002, 40, 754–760. [Google Scholar] [CrossRef]
- Perregaux, D.; Chaudhuri, A.; Rao, S.; Airen, A.; Wilson, M.; Sung, B.H.; Dandona, P. Brachial vascular reactivity in blacks. Hypertension 2000, 36, 866–871. [Google Scholar] [CrossRef]
- Feairheller, D.L.; Park, J.Y.; Sturgeon, K.M.; Williamson, S.T.; Diaz, K.M.; Veerabhadrappa, P.; Brown, M.D. Racial differences in oxidative stress and inflammation: In vitro and in vivo. Clin. Transl. Sci. 2011, 4, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Feairheller, D.L.; Thakkar, S.; Veerabhadrappa, P.; Park, J.Y. Racial differences in tumor necrosis factor-α-induced endothelial microparticles and interleukin-6 production. Vasc. Health Risk Manag. 2011, 7, 541–550. [Google Scholar] [CrossRef]
- Jones, D.S.; Andrawis, N.S.; Abernethy, D.R. Impaired endothelial-dependent forearm vascular relaxation in black Americans. Clin. Pharmacol. Ther. 1999, 65, 408–412. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, R.B., Jr.; Burke, G.; O’Leary, D.; Rewers, M.; Selby, J.; Savage, P.J.; Saad, M.F.; Bergman, R.N.; Howard, G.; Wagenknecht, L.; et al. Ethnic differences in carotid wall thickness. The Insulin Resistance Atherosclerosis Study. Stroke 1996, 27, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Lange, L.A.; Bowden, D.W.; Langefeld, C.D.; Wagenknecht, L.E.; Carr, J.J.; Rich, S.S.; Riley, W.A.; Freedman, B.I. Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke 2002, 33, 1876–1881. [Google Scholar] [CrossRef]
- Skoog, T.; Dichtl, W.; Boquist, S.; Skoglund-Andersson, C.; Karpe, F.; Tang, R.; Bond, M.G.; de Faire, U.; Nilsson, J.; Eriksson, P.; et al. Plasma tumour necrosis factor-alpha and early carotid atherosclerosis in healthy middle-aged men. Eur. Heart J. 2002, 23, 376–383. [Google Scholar] [CrossRef]
- Hwang, S.J.; Ballantyne, C.M.; Sharrett, A.R.; Smith, L.C.; Davis, C.E.; Gotto, A.M., Jr.; Boerwinkle, E. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: The Atherosclerosis Risk In Communities (ARIC) study. Circulation 1997, 96, 4219–4225. [Google Scholar] [CrossRef]
- Cook, M.D.; Ling, C.; Grimm, H.; Adeyemo, A.; Aldokhayyil, M.; Heffernan, K.; Fernhall, B.; Brown, M.J.A.R. Primary African American Endothelial Cells Exhibit Endothelial Dysfunction with an Exacerbated Inflammatory Profile and Blunted MMP-2 Activity. Artery Res. 2020, 27, 38–46. [Google Scholar] [CrossRef]
- Qin, Z. The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis 2012, 221, 2–11. [Google Scholar] [CrossRef]
- Deo, S.H.; Holwerda, S.W.; Keller, D.M.; Fadel, P.J. Elevated peripheral blood mononuclear cell-derived superoxide production in healthy young black men. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H548–H552. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Scheurich, P. Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int. J. Biochem. Cell Biol. 2001, 33, 19–32. [Google Scholar] [CrossRef]
- Pereira, D.A.; Ribeiro-Samora, G.A.; Vieira, D.S.; Pereira, L.S.; Coelho, F.M.; Parreira, V.F.; Moreira Mda, C.; Alencar, M.C.; Britto, R.R. Evaluation of the inflammatory response to two different intensities of exercise in individuals with heart failure. Inflammation 2012, 35, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R.; Pober, J.S. Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 2001, 20, 6482–6491. [Google Scholar] [CrossRef]
- Chung, J.Y.; Park, Y.C.; Ye, H.; Wu, H. All TRAFs are not created equal: Common and distinct molecular mechanisms of TRAF-mediated signal transduction. J. Cell Sci. 2002, 115 Pt 4, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Häcker, H.; Tseng, P.H.; Karin, M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat. Rev. Immunol. 2011, 11, 457–468. [Google Scholar] [CrossRef]
- Gissler, M.C.; Stachon, P.; Wolf, D.; Marchini, T. The Role of Tumor Necrosis Factor Associated Factors (TRAFs) in Vascular Inflammation and Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 826630. [Google Scholar] [CrossRef]
- He, J.Q.; Oganesyan, G.; Saha, S.K.; Zarnegar, B.; Cheng, G. TRAF3 and its biological function. Adv. Exp. Med. Biol. 2007, 597, 48–59. [Google Scholar] [CrossRef]
- Bishop, G.A.; Stunz, L.L.; Hostager, B.S. TRAF3 as a Multifaceted Regulator of B Lymphocyte Survival and Activation. Front. Immunol. 2018, 9, 2161. [Google Scholar] [CrossRef]
- Au, P.Y.; Yeh, W.C. Physiological roles and mechanisms of signaling by TRAF2 and TRAF5. Adv. Exp. Med. Biol. 2007, 597, 32–47. [Google Scholar] [CrossRef]
- Urbich, C.; Mallat, Z.; Tedgui, A.; Clauss, M.; Zeiher, A.M.; Dimmeler, S. Upregulation of TRAF-3 by shear stress blocks CD40-mediated endothelial activation. J. Clin. Investig. 2001, 108, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Missiou, A.; Rudolf, P.; Stachon, P.; Wolf, D.; Varo, N.; Aichele, P.; Colberg, C.; Hoppe, N.; Ernst, S.; Münkel, C.; et al. TRAF5 deficiency accelerates atherogenesis in mice by increasing inflammatory cell recruitment and foam cell formation. Circ. Res. 2010, 107, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Russell, F.D.; Hamilton, K.D. Nutrient deprivation increases vulnerability of endothelial cells to proinflammatory insults. Free Radic. Biol. Med. 2014, 67, 408–415. [Google Scholar] [CrossRef]
- Luu, N.T.; Rahman, M.; Stone, P.C.; Rainger, G.E.; Nash, G.B. Responses of endothelial cells from different vessels to inflammatory cytokines and shear stress: Evidence for the pliability of endothelial phenotype. J. Vasc. Res. 2010, 47, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Feairheller, D.L.; Park, J.Y.; Rizzo, V.; Kim, B.; Brown, M.D. Racial differences in the responses to shear stress in human umbilical vein endothelial cells. Vasc. Health Risk Manag. 2011, 7, 425–431. [Google Scholar] [CrossRef]
- Kalinowski, L.; Dobrucki, I.T.; Malinski, T. Race-specific differences in endothelial function: Predisposition of African Americans to vascular diseases. Circulation 2004, 109, 2511–2517. [Google Scholar] [CrossRef]
- Robinson, A.T.; Cook, M.D.; Lane-Cordova, A.D. Making cell culture more physiological: A call for a more comprehensive assessment of racial disparities in endothelial cell culture studies. Am. J. Physiol. Cell Physiol. 2020, 318, C238–C241. [Google Scholar] [CrossRef]
- Dewey, C.F., Jr.; Bussolari, S.R.; Gimbrone, M.A., Jr.; Davies, P.F. The dynamic response of vascular endothelial cells to fluid shear stress. J. Biomech. Eng. 1981, 103, 177–185. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldokhayyil, M.; Gomez, D.H.; Cook, M.D.; Kavazis, A.N.; Roberts, M.D.; Geetha, T.; Brown, M.D. Influence of Race and High Laminar Shear Stress on TNFR1 Signaling in Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 14723. https://doi.org/10.3390/ijms241914723
Aldokhayyil M, Gomez DH, Cook MD, Kavazis AN, Roberts MD, Geetha T, Brown MD. Influence of Race and High Laminar Shear Stress on TNFR1 Signaling in Endothelial Cells. International Journal of Molecular Sciences. 2023; 24(19):14723. https://doi.org/10.3390/ijms241914723
Chicago/Turabian StyleAldokhayyil, Maitha, Dulce H. Gomez, Marc D. Cook, Andreas N. Kavazis, Michael D. Roberts, Thangiah Geetha, and Michael D. Brown. 2023. "Influence of Race and High Laminar Shear Stress on TNFR1 Signaling in Endothelial Cells" International Journal of Molecular Sciences 24, no. 19: 14723. https://doi.org/10.3390/ijms241914723