
β-Lactam TRPM8 Antagonists Derived from Phe-Phenylalaninol Conjugates: Structure–Activity Relationships and Antiallodynic Activity

 , , , , , ,
, , , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
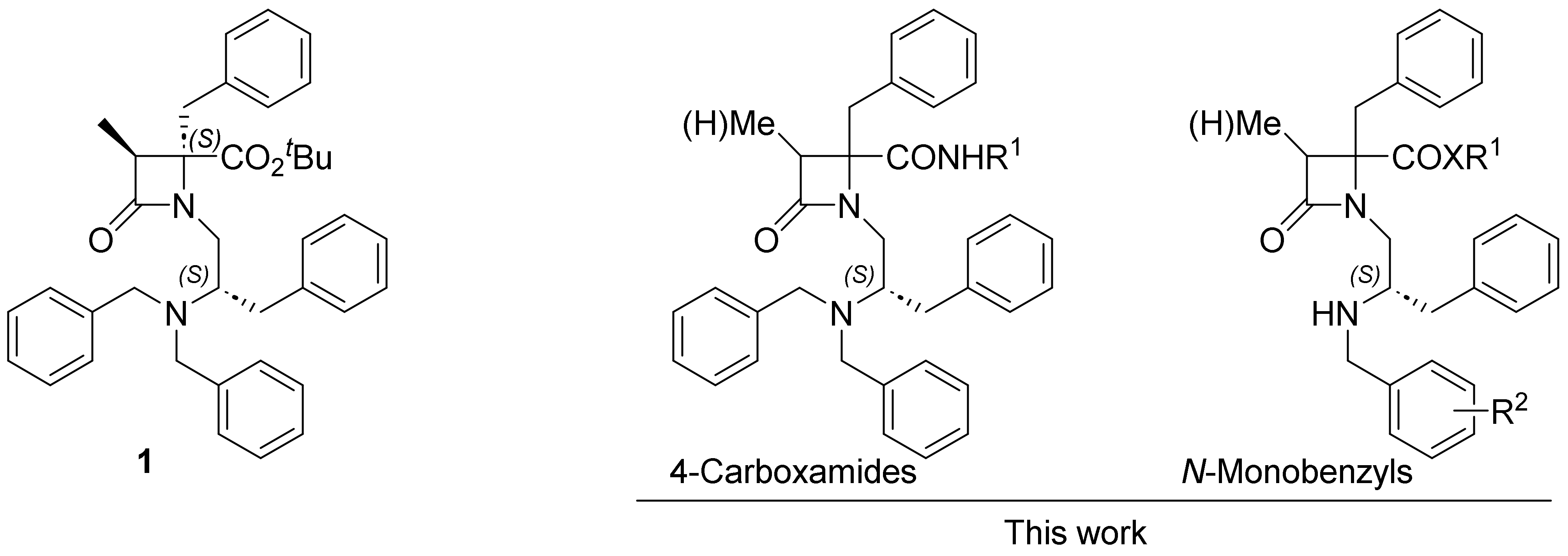
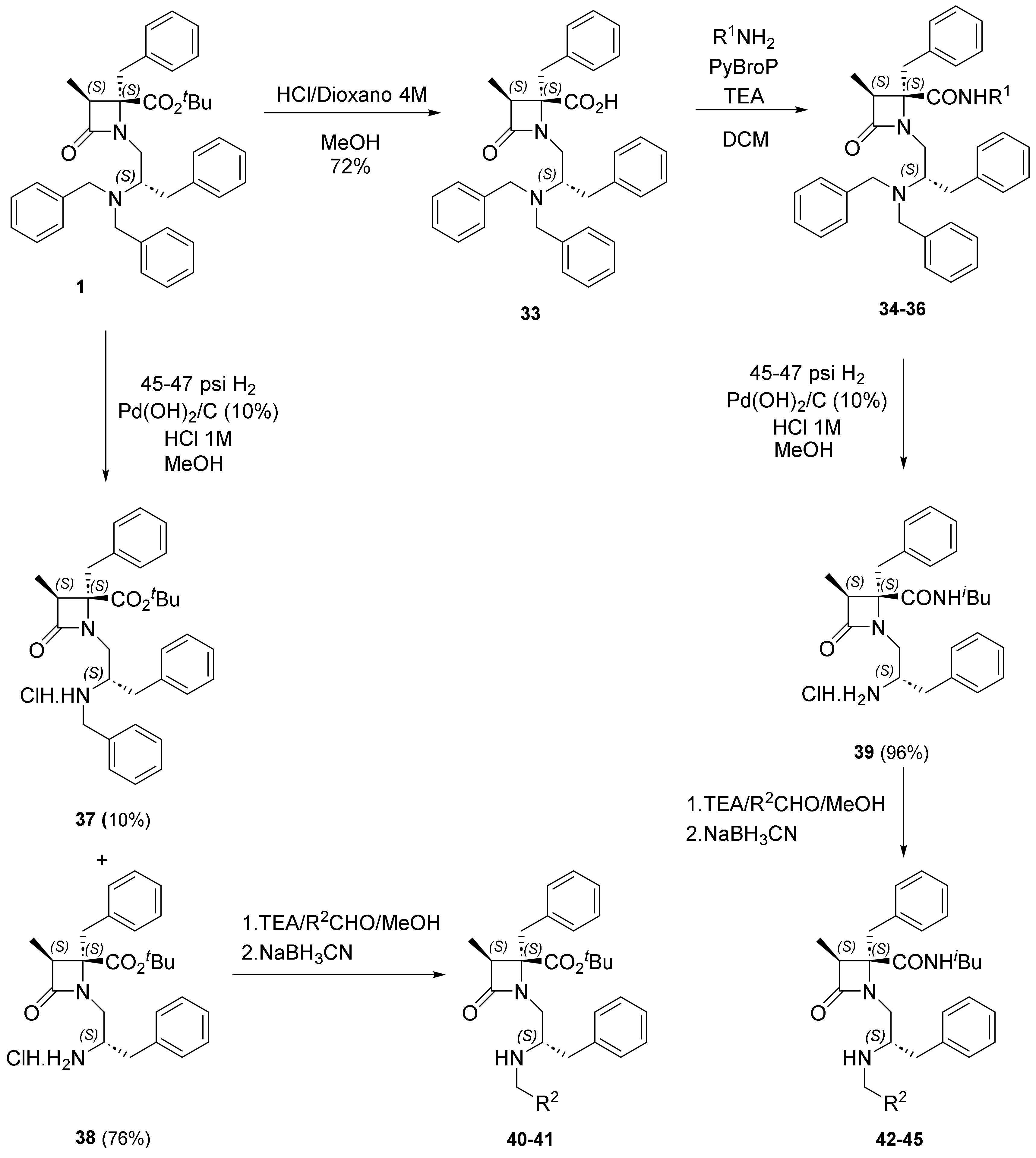
2.1. 1,4,4-Trisubstituted β-Lactam Derivatives
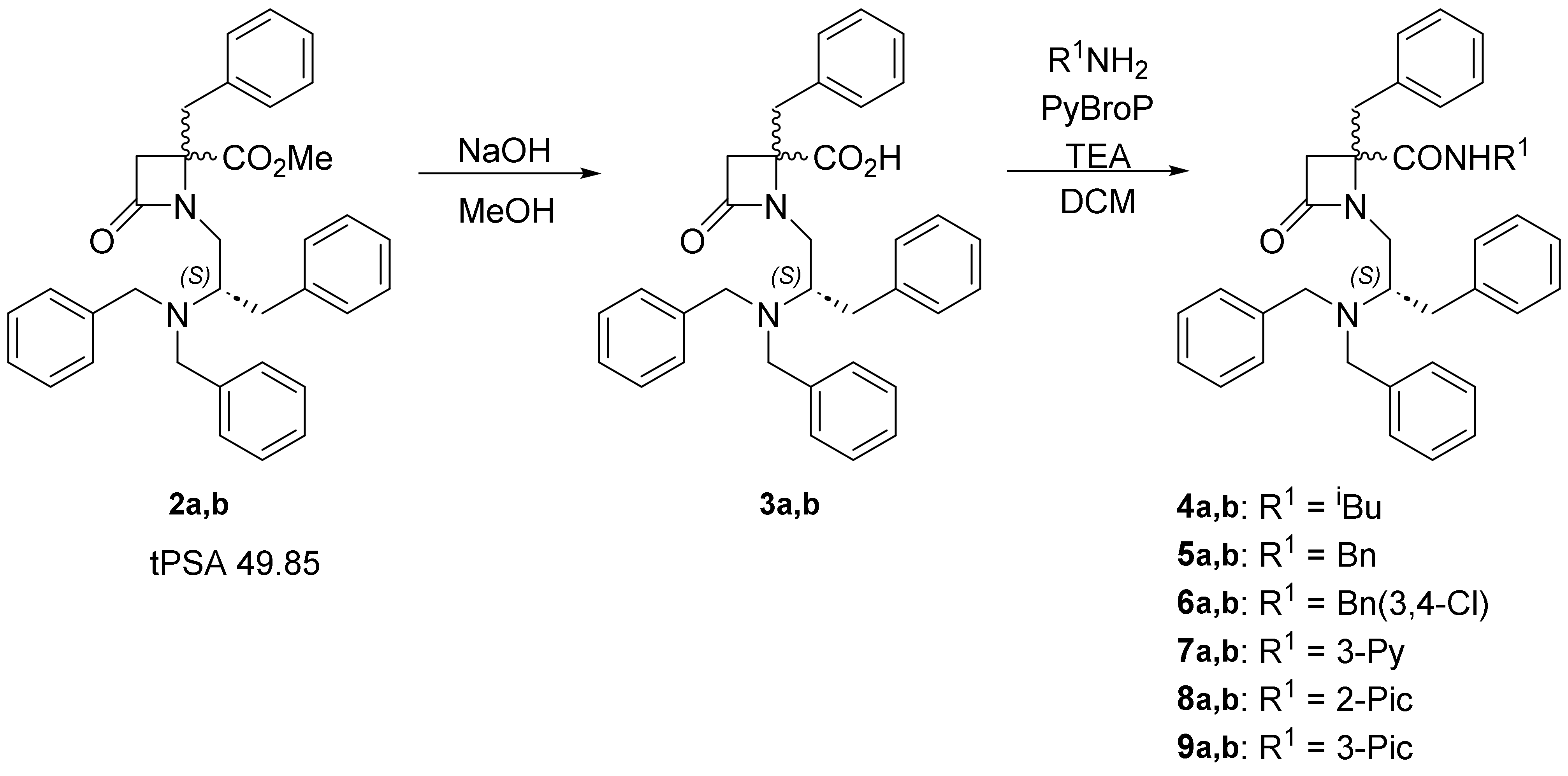
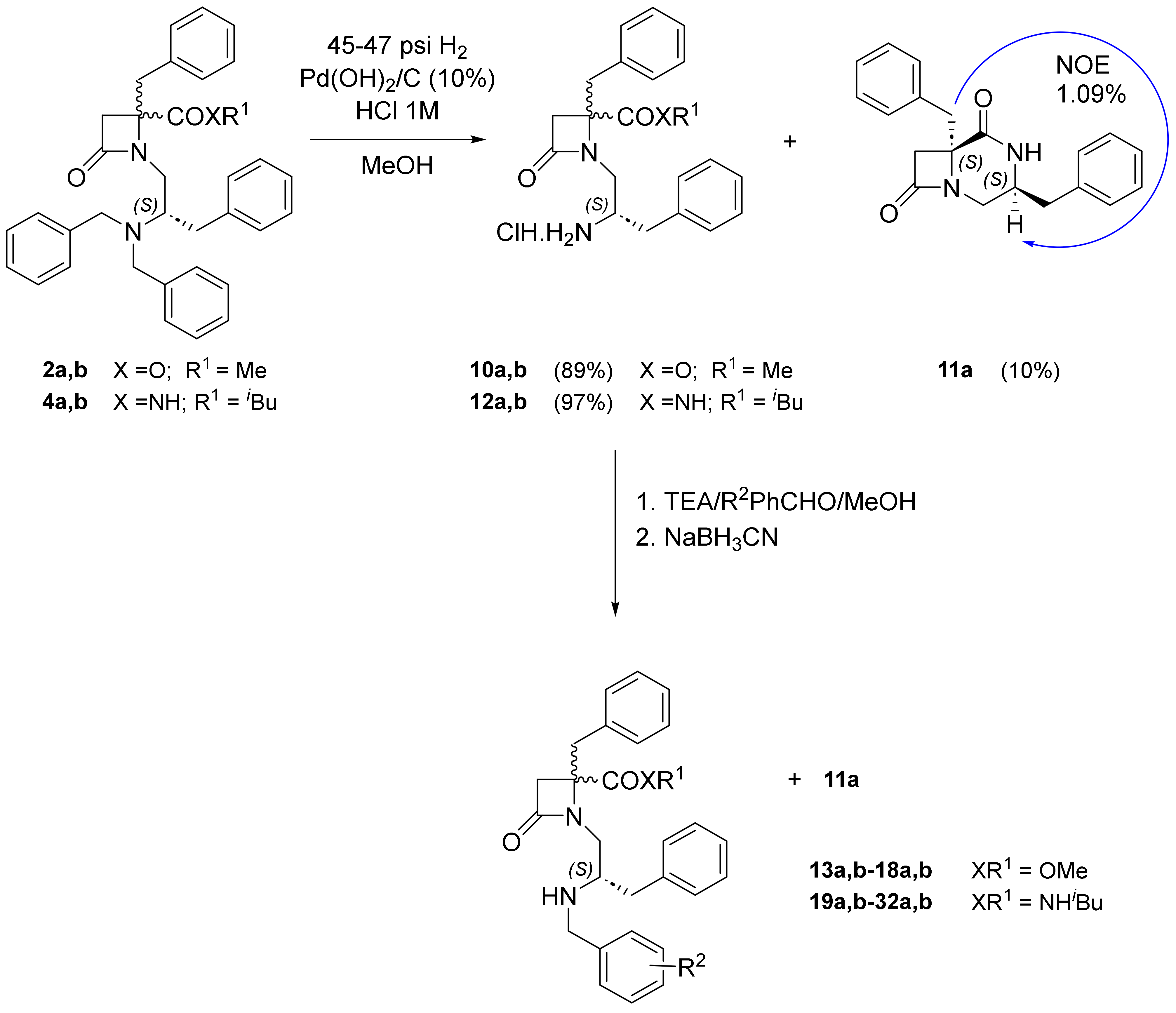
2.2. 1,3,4,4-Tetrasubstituted, Enantiopure β-Lactam Derivatives
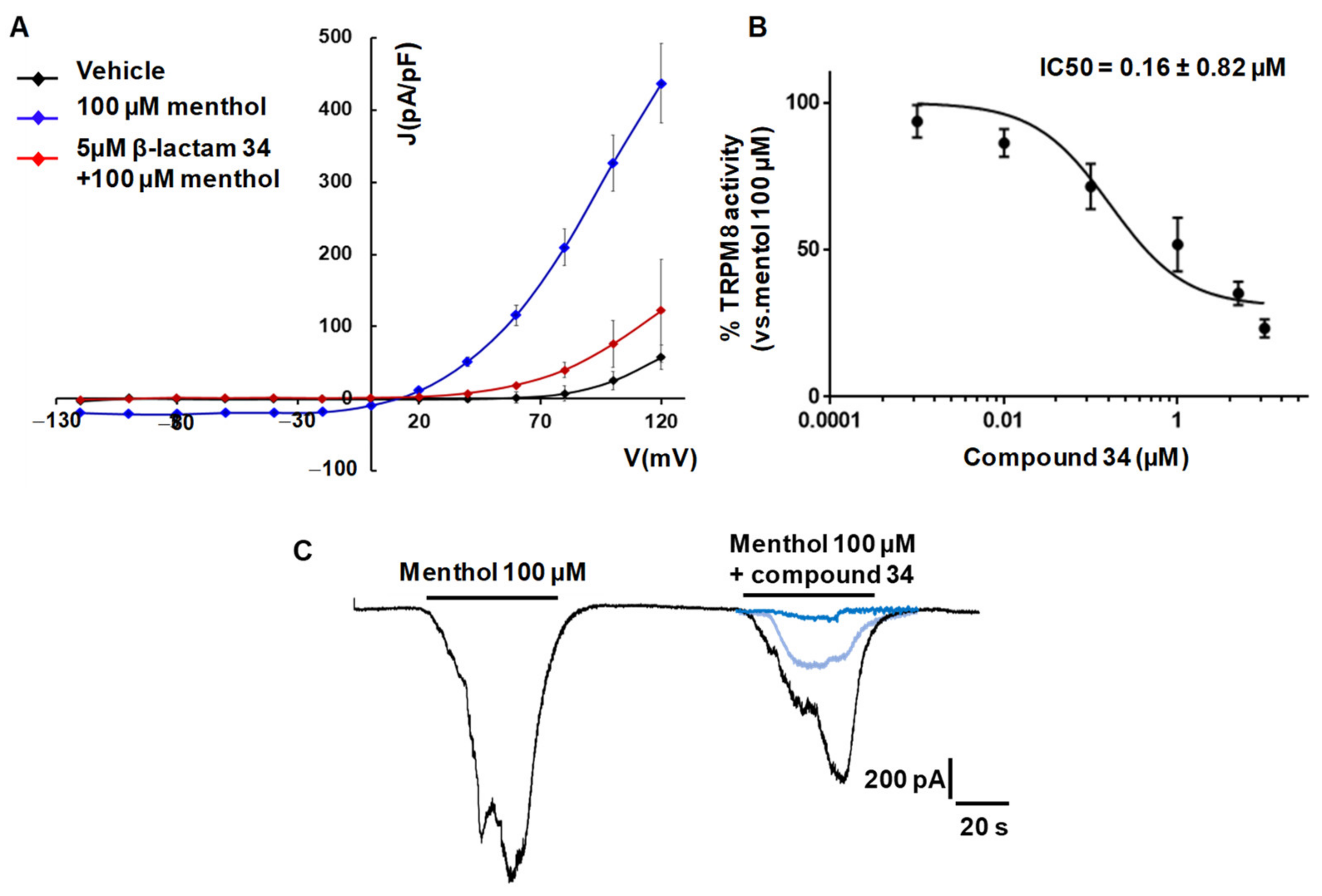
2.3. Activity in hTRPM8 Channels and Electrophysiology Assays
2.4. Activity in Other TRP Channels and Pain-Related Peripheral Receptors
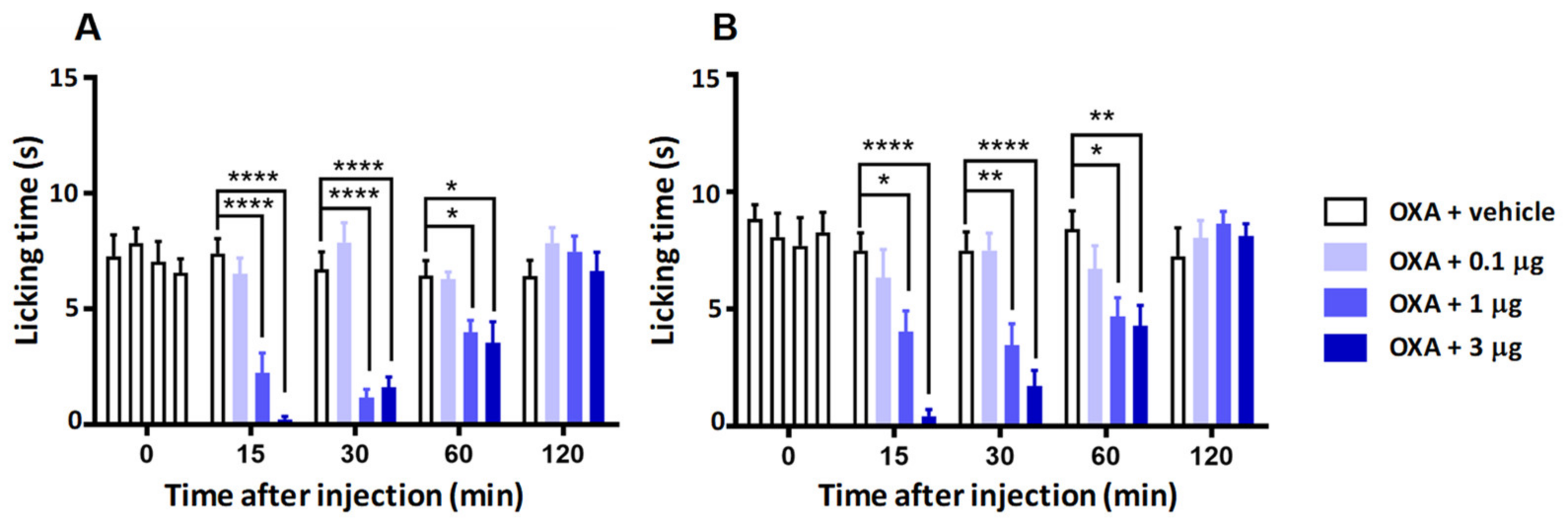
2.5. Antinociceptive Activity in a Mouse Model of Chemotherapy-Induced Cold Allodynia
2.6. Insights into the Mode of Interaction of Selected β-Lactams with the TRPM8 Channel
3. Materials and Methods
3.1. Synthesis
3.2. Molecular Modeling
3.3. Biological Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kashio, M.; Tominaga, M. TRP channels in thermosensation. Curr. Opin. Neurobiol. 2022, 75, 102591. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M. Temperature sensing. Igaku No Ayumi 2019, 270, 998–1003. [Google Scholar]
- Yue, L.; Xu, H. TRP channels in health and disease at a glance. J. Cell Sci. 2021, 134, jcs258372. [Google Scholar] [CrossRef] [PubMed]
- Guibert, C.; Ducret, T.; Savineau, J.-P. Expression and physiological roles of TRP channels in smooth muscle cells. Adv. Exp. Med. Biol. 2011, 704, 687–706. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Vriens, J.; Vennekens, R. Targeting TRP Channels—Valuable Alternatives to Combat Pain, Lower Urinary Tract Disorders, and Type 2 Diabetes? Trends Pharmacol. Sci. 2019, 40, 669–683. [Google Scholar] [CrossRef]
- Gonzalez-Cobos, J.C.; Zhang, X.; Motiani, R.K.; Harmon, K.E.; Trebak, M. TRPs to Cardiovascular Disease; Springer: Berlin/Heidelberg, Germany, 2012; Volume 2, pp. 3–40. [Google Scholar]
- Perez de Vega, M.J.; Gomez-Monterrey, I.; Ferrer-Montiel, A.; Gonzalez-Muniz, R. Transient Receptor Potential Melastatin 8 Channel (TRPM8) Modulation: Cool Entryway for Treating Pain and Cancer. J. Med. Chem. 2016, 59, 10006–10029. [Google Scholar] [CrossRef]
- Izquierdo, C.; Martin-Martinez, M.; Gomez-Monterrey, I.; Gonzalez-Muniz, R. TRPM8 Channels: Advances in Structural Studies and Pharmacological Modulation. Int. J. Mol. Sci. 2021, 22, 8502. [Google Scholar] [CrossRef]
- Zhang, L.; Barritt, G.J. TRPM8 in prostate cancer cells: A potential diagnostic and prognostic marker with a secretory function? Endocr. Relat. Cancer 2006, 13, 27–38. [Google Scholar] [CrossRef]
- Bautista, D.M.; Siemens, J.; Glazer, J.M.; Tsuruda, P.R.; Basbaum, A.I.; Stucky, C.L.; Jordt, S.E.; Julius, D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007, 448, 204–208. [Google Scholar] [CrossRef]
- Dhaka, A.; Earley, T.J.; Watson, J.; Patapoutian, A. Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J. Neurosci. 2008, 28, 566–575. [Google Scholar] [CrossRef]
- De Caro, C.; Cristiano, C.; Avagliano, C.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Gomez-Monterrey, I.; La Rana, G.; Gualillo, O.; Calignano, A.; et al. Characterization of new TRPM8 modulators in pain perception. Int. J. Mol. Sci. 2019, 20, 5544. [Google Scholar] [CrossRef]
- Soeda, M.; Ohka, S.; Nishizawa, D.; Hasegawa, J.; Nakayama, K.; Ebata, Y.; Ikeda, K.; Soeda, M.; Fukuda, K.-I.; Ichinohe, T. Cold pain sensitivity is associated with single-nucleotide polymorphisms of PAR2/F2RL1 and TRPM8. Mol. Pain 2021, 17, 17448069211002008. [Google Scholar] [CrossRef] [PubMed]
- Pertusa, M.; Solorza, J.; Madrid, R. Molecular determinants of TRPM8 function: Key clues for a cool modulation. Front. Pharmacol. 2023, 14, 1213337. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Owsianik, G.; Nilius, B. TRPM8. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2007; Volume 179, pp. 329–344. [Google Scholar] [CrossRef]
- Almaraz, L.; Manenschijn, J.-A.; de la Pena, E.; Viana, F. TRPM8. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 222, pp. 547–579. [Google Scholar] [CrossRef]
- Liu, Y.; Mikrani, R.; He, Y.; Faran Ashraf Baig, M.M.; Abbas, M.; Naveed, M.; Tang, M.; Zhang, Q.; Li, C.; Zhou, X. TRPM8 channels: A review of distribution and clinical role. Eur. J. Pharmacol. 2020, 882, 173312. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, N.; Fujita, T. The TRPM8 channel as a potential therapeutic target for bladder hypersensitive disorders. J. Smooth Muscle Res. 2022, 58, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Su, X.; Zhang, W.; Zhang, Y.-H.; Feng, X.; Ji, Y.-H.; Tan, Z.-Y. Oxaliplatin depolarizes the IB4- dorsal root ganglion neurons to drive the development of neuropathic pain through TRPM8 in mice. Front. Mol. Neurosci. 2021, 14, 690858. [Google Scholar] [CrossRef]
- Aierken, A.; Xie, Y.-K.; Dong, W.; Apaer, A.; Lin, J.-J.; Zhao, Z.; Yang, S.; Xu, Z.-Z.; Yang, F. Rational Design of a Modality-Specific Inhibitor of TRPM8 Channel against Oxaliplatin-Induced Cold Allodynia. Adv. Sci. 2021, 8, 2101717. [Google Scholar] [CrossRef]
- Fakih, D.; Baudouin, C.; Goazigo, A.R.-L.; Parsadaniantz, S.M. TRPM8: A therapeutic target for neuroinflammatory symptoms induced by severe dry eye disease. Int. J. Mol. Sci. 2020, 21, 8756. [Google Scholar] [CrossRef]
- Liu, X.-R.; Liu, Q.; Chen, G.-Y.; Hu, Y.; Sham, J.S.K.; Lin, M.-J. Down-Regulation of TRPM8 in Pulmonary Arteries of Pulmonary Hypertensive Rats. Cell. Physiol. Biochem. 2013, 31, 892–904. [Google Scholar] [CrossRef]
- Naumov, D.E.; Kotova, O.O.; Gassan, D.A.; Sugaylo, I.Y.; Afanas’eva, E.Y.; Sheludko, E.G.; Perelman, J.M. Effect of TRPM8 and TRPA1 Polymorphisms on COPD Predisposition and Lung Function in COPD Patients. J. Pers. Med. 2021, 11, 108. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Q.; Hua, L.; Pan, J. Inhibition of transient receptor potential melastatin 8 alleviates airway inflammation and remodeling in a murine model of asthma with cold air stimulus. Acta Biochim. Biophys. Sin. 2018, 50, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Szallasi, A. ThermoTRP Channel Expression in Cancers: Implications for Diagnosis and Prognosis (Practical Approach by a Pathologist). Int. J. Mol. Sci. 2023, 24, 9098. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Muniz, R.; Bonache, M.A.; Martin-Escura, C.; Gomez-Monterrey, I. Recent progress in TRPM8 modulation: An update. Int. J. Mol. Sci. 2019, 20, 2618. [Google Scholar] [CrossRef] [PubMed]
- Horne, D.B.; Biswas, K.; Brown, J.; Bartberger, M.D.; Clarine, J.; Davis, C.D.; Gore, V.K.; Harried, S.; Horner, M.; Kaller, M.R.; et al. Discovery of TRPM8 Antagonist (S)-6-(((3-Fluoro-4-(trifluoromethoxy)phenyl)(3-fluoropyridin-2-yl)methyl)carbamoyl)nicotinic Acid (AMG 333), a Clinical Candidate for the Treatment of Migraine. J. Med. Chem. 2018, 61, 8186–8201. [Google Scholar] [CrossRef]
- Andrews, M.D.; Af Forselles, K.; Beaumont, K.; Galan, S.R.G.; Glossop, P.A.; Grenie, M.; Jessiman, A.; Kenyon, A.S.; Lunn, G.; Maw, G.; et al. Discovery of a selective TRPM8 antagonist with clinical efficacy in cold-related pain. ACS Med. Chem. Lett. 2015, 6, 419–424. [Google Scholar] [CrossRef]
- Fernandez-Carvajal, A.; Gonzalez-Muniz, R.; Fernandez-Ballester, G.; Ferrer-Montiel, A. Investigational drugs in early phase clinical trials targeting thermotransient receptor potential (thermoTRP) channels. Expert Opin. Investig. Drugs 2020, 29, 1209–1222. [Google Scholar] [CrossRef]
- Gosset, J.R.; Beaumont, K.; Matsuura, T.; Winchester, W.; Attkins, N.; Glatt, S.; Lightbown, I.; Ulrich, K.; Roberts, S.; Harris, J.; et al. A cross-species translational pharmacokinetic-pharmacodynamic evaluation of core body temperature reduction by the TRPM8 blocker PF-05105679. Eur. J. Pharm. Sci. 2017, 109S, S161–S167. [Google Scholar] [CrossRef]
- Gavva, N.R.; Davis, C.; Lehto, S.G.; Rao, S.; Wang, W.; Zhu, D.X.D. Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol. Pain 2012, 8, 115. [Google Scholar] [CrossRef]
- Thapa, D.; Barrett, B.; Argunhan, F.; Brain, S.D. Influence of Cold-TRP Receptors on Cold-Influenced Behaviour. Pharmaceuticals 2022, 15, 42. [Google Scholar] [CrossRef]
- de la Torre-Martinez, R.; Bonache, M.A.; Llabres-Campaner, P.J.; Balsera, B.; Fernandez-Carvajal, A.; Fernandez-Ballester, G.; Ferrer-Montiel, A.; Perez de Vega, M.J.; Gonzalez-Muniz, R. Synthesis, high-throughput screening and pharmacological characterization of β-lactam derivatives as TRPM8 antagonists. Sci. Rep. 2017, 7, 10766. [Google Scholar] [CrossRef]
- Bonache, M.Á.; Llabrés, P.J.; Martín-Escura, C.; De la Torre-Martínez, R.; Medina-Peris, A.; Butrón, L.; Gómez-Monterrey, I.; Roa, A.M.; Fernández-Ballester, G.; Ferrer-Montiel, A.; et al. Phenylalanine-derived β-lactam trpm8 modulators. Configuration effect on the antagonist activity. Int. J. Mol. Sci. 2021, 22, 2370. [Google Scholar] [CrossRef]
- Martin-Escura, C.; Medina-Peris, A.; Spear, L.A.; de la Torre Martinez, R.; Olivos-Ore, L.A.; Barahona, M.V.; Gonzalez-Rodriguez, S.; Fernandez-Ballester, G.; Fernandez-Carvajal, A.; Artalejo, A.R.; et al. β-Lactam TRPM8 Antagonist RGM8-51 Displays Antinociceptive Activity in Different Animal Models. Int. J. Mol. Sci. 2022, 23, 2692. [Google Scholar] [CrossRef] [PubMed]
- Bonache, M.A.; Martin-Escura, C.; de la Torre Martinez, R.; Medina, A.; Gonzalez-Rodriguez, S.; Francesch, A.; Cuevas, C.; Roa, A.M.; Fernandez-Ballester, G.; Ferrer-Montiel, A.; et al. Highly functionalized β-lactams and 2-ketopiperazines as TRPM8 antagonists with antiallodynic activity. Sci. Rep. 2020, 10, 14154. [Google Scholar] [CrossRef] [PubMed]
- Werkheiser, J.L.; Rawls, S.M.; Cowan, A. Mu and kappa opioid receptor agonists antagonize icilin-induced wet-dog shaking in rats. Eur. J. Pharmacol. 2006, 547, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Perez-Faginas, P.; O’Reilly, F.; O’Byrne, A.; Garcia-Aparicio, C.; Martin-Martinez, M.; Perez de Vega, M.J.; Garcia-Lopez, M.T.; Gonzalez-Muniz, R. Exceptional Stereoselectivity in the Synthesis of 1,3,4-Trisubstituted 4-Carboxy β-Lactam Derivatives from Amino Acids. Org. Lett. 2007, 9, 1593–1596. [Google Scholar] [CrossRef]
- Journigan, V.B.; Alarcón-Alarcón, D.; Feng, Z.; Wang, Y.; Liang, T.; Dawley, D.C.; Amin, A.R.M.R.; Montano, C.; Van Horn, W.D.; Xie, X.Q.; et al. Structural and in Vitro Functional Characterization of a Menthyl TRPM8 Antagonist Indicates Species-Dependent Regulation. ACS Med. Chem. Lett. 2021, 12, 758–767. [Google Scholar] [CrossRef]
- Xu, L.; Han, Y.; Chen, X.; Aierken, A.; Wen, H.; Zheng, W.; Wang, H.; Lu, X.; Zhao, Z.; Ma, C.; et al. Molecular mechanisms underlying menthol binding and activation of TRPM8 ion channel. Nat. Commun. 2020, 11, 3790. [Google Scholar] [CrossRef]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.-Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, eaav9334. [Google Scholar] [CrossRef]
- Vangeel, L.; Benoit, M.; Miron, Y.; Miller, P.E.; De Clercq, K.; Chaltin, P.; Verfaillie, C.; Vriens, J.; Voets, T. Functional expression and pharmacological modulation of TRPM3 in human sensory neurons. Br. J. Pharmacol. 2020, 177, 2683–2695. [Google Scholar] [CrossRef]
- Li, W.G.; Xu, T. Le ASIC3 channels in multimodal sensory perception. ACS Chem. Neurosci. 2011, 2, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Khan, S.; Kim, Y.S. Insight into Pain Modulation: Nociceptors Sensitization and Therapeutic Targets. Curr. Drug Targets 2019, 20, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yamaki, S.; Jung, T.; Kim, B.; Huyhn, R.; McKemy, D.D. Endogenous inflammatory mediators produced by injury activate TRPV1 and TRPA1 nociceptors to induce sexually dimorphic cold pain that is dependent on TRPM8 and GFRa3. J. Neurosci. 2023, 43, 2803–2814. [Google Scholar] [CrossRef]
- Chen, S.R.; Chen, H.; Yuan, W.X.; Wess, J.; Pan, H.L. Dynamic control of glutamatergic synaptic input in the spinal cord by muscarinic receptor subtypes defined using knockout mice. J. Biol. Chem. 2010, 285, 40427–40437. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Go, D.; Kim, W.; Lee, G.; Bae, H.; Quan, F.S.; Kim, S.K. Involvement of spinal muscarinic and serotonergic receptors in the anti-allodynic effect of electroacupuncture in rats with oxaliplatin-induced neuropathic pain. Korean J. Physiol. Pharmacol. 2016, 20, 407–414. [Google Scholar] [CrossRef]
- Camilleri, M. Toward an effective peripheral visceral analgesic: Responding to the national opioid crisis. Am. J. Physiol. 2018, 314, G637. [Google Scholar] [CrossRef]
- Beijers, A.J.M.; Jongen, J.L.M.; Vreugdenhil, G. Chemotherapy-induced neurotoxicity: The value of neuroprotective strategies. Neth. J. Med. 2012, 70, 18–25. [Google Scholar]
- Rimola, V.; Osthues, T.; Koenigs, V.; Geisslinger, G.; Sisignano, M. Oxaliplatin causes transient changes in TRPM8 channel activity. Int. J. Mol. Sci. 2021, 22, 4962. [Google Scholar] [CrossRef]
- Journigan, V.B.; Feng, Z.; Rahman, S.; Wang, Y.; Amin, A.R.M.R.; Heffner, C.E.; Bachtel, N.; Wang, S.; Gonzalez-Rodriguez, S.; Fernández-Carvajal, A.; et al. Structure-Based Design of Novel Biphenyl Amide Antagonists of Human Transient Receptor Potential Cation Channel Subfamily M Member 8 Channels with Potential Implications in the Treatment of Sensory Neuropathies. ACS Chem. Neurosci. 2020, 11, 268–290. [Google Scholar] [CrossRef]
- Bertamino, A.; Ostacolo, C.; Medina, A.; Di Sarno, V.; Lauro, G.; Ciaglia, T.; Vestuto, V.; Pepe, G.; Basilicata, M.G.; Musella, S.; et al. Exploration of TRPM8 Binding Sites by β-Carboline-Based Antagonists and Their In Vitro Characterization and In Vivo Analgesic Activities. J. Med. Chem. 2020, 63, 9672–9694. [Google Scholar] [CrossRef]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.-Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science 2018, 359, 237–241. [Google Scholar] [CrossRef]
- Diver, M.M.; Cheng, Y.; Julius, D. Structural insights into TRPM8 inhibition and desensitization. Science 2019, 365, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- McCoy, D.D.; Palkar, R.; Yang, Y.; Ongun, S.; McKemy, D.D. Cellular permeation of large molecules mediated by TRPM8 channels. Neurosci. Lett. 2017, 639, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock and AutoDockTools: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Ozvoldik, K.; Stockner, T.; Rammner, B.; Krieger, E. Assembly of Biomolecular Gigastructures and Visualization with the Vulkan Graphics API. J. Chem. Inf. Model. 2021, 61, 5293–5303. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443. [Google Scholar] [CrossRef]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.-D.; Benoit, M.; Xue, F.-Q.; Janssens, A.; Kerselaers, S.; et al. TRPM3 Is a Nociceptor Channel Involved in the Detection of Noxious Heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef]
- González-Muñiz, R.; Pérez de Vega, M.J.; Bonache de Marcos, M.Á.; Ferrer-Montiel, A.; Fernández-Carvajal, A.; de la Torre-Martinez, R. Heterocyclic Compounds as TRPM8 Channel Antagonists and Uses Thereof. WO 2017005950 12 January 2017. Available online: http://hdl.handle.net/10261/176227 (accessed on 1 September 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | a:b | IC50 (μM) | 95% Confidential Intervals |
|---|---|---|---|---|
| 2a,b | 1:1.8 | 0.15 ± 1.23 | 0.095 to 0.22 | |
| 4a,b | iBu | 1:1.6 | 0.71 ± 1.20 | 0.49 to 1.03 |
| 5a,b | Bn | 1:2.6 | 0.71 ± 1.27 | 0.43 to 1.16 |
| 6a | (3,4-Cl)Bn | ND | - | |
| 6a,b | (3,4-Cl)Bn | 1:1.4 | ND | - |
| 7a,b | 3-Py | 1:1.4 | 11.62 ± 1.30 | 6.73 to 20.06 |
| 8a,b | 2-Pic | 1:1.8 | 1.99 ± 1.26 | 1.24 to 3.17 |
| 9a,b | 3-Pic | 1:1.2 | 10.69 ± 1.38 | 5.52 to 20.73 |
| 1 | 1.06 ± 1.21 | 0.72 to 1.55 | ||
| AMTB | 7.3 ± 1.5 | 8.82 to 7.85 |
| Compound | XR1 | R2 | a:b | IC50 (μM) | 95% Confidential Intervals |
|---|---|---|---|---|---|
| 2a,b | OMe | - | 1:1.8 | 0.15 ± 1.23 | 0.095 to 0.22 |
| 4a,b | NHiBu | - | 1:1.6 | 0.71 ± 1.20 | 0.49 to 1.03 |
| 11a | - | - | - | ND | ND |
| 13a,b | OMe | H | 1:3.6 | 3.95 ± 1.20 | 2.66 to 5.85 |
| 14a,b | OMe | 3-Me | 1:3.6 | 3.58 ± 1.41 | 1.74 to 7.35 |
| 15a,b | OMe | 4-Me | 1:5.6 | 3.90 ± 1.47 | 1.74 to 8.74 |
| 16a,b | OMe | 3-Br | 1:2 | 1.10 ± 1.38 | 0.56 to 2.16 |
| 17a,b | OMe | (2-Nph)CH2 | 1:4 | 2.60 ± 1.23 | 1.69 to 3.98 |
| 18a,b | OMe | 4-OPr | 1:9.5 | 8.90 ± 1.40 | 4.37 to 18.10 |
| 19a,b | NHiBu | 3-Me | 1:1.5 | 3.97 ± 1.31 | 2.28 to 6.93 |
| 20a,b | NHiBu | 3-Ph | 1:1.1 | 2.03 ± 1.32 | 1.15 to 3.60 |
| 21a,b | NHiBu | 3-F | 1:2.6 | 5.15 ± 1.46 | 2.37 to 11.22 |
| 22a,b | NHiBu | 3-Cl | 1:2.4 | 2.57 ± 1.32 | 1.45 to 4.53 |
| 23a,b | NHiBu | 3-Br | 1:2.9 | 3.30 ± 1.36 | 1.73 to 6.29 |
| 24a,b | NHiBu | 3-I | 1:2.7 | 3.83 ± 1.28 | 2.33 to 6.29 |
| 25a,b | NHiBu | 3-OMe | 1:2.5 | 4.48 ± 1.25 | 2.83 to 7.09 |
| 26a,b | NHiBu | 3-OPh | 1:5.7 | 8.22 ± 1.30 | 4.79 to 14.08 |
| 27a,b | NHiBu | 3-OBn | 1:1.5 | 3.27 ± 1.37 | 1.72 to 6.20 |
| 28a,b | NHiBu | 3-CN | 1:1.1 | 12.10 ± 1.31 | 6.99 to 20.94 |
| 29a,b | NHiBu | 3-NO2 | 1.2:1 | 2.12 ± 1.28 | 1.28 to 3.50 |
| 30a,b | NHiBu | 4-Ph | 1:1.9 | 4.57 ± 1.22 | 3.07 to 6.82 |
| 31a,b | NHiBu | 4-F | 1.1:1 | 8.32 ± 1.30 | 4.87 to 14.22 |
| 32a,b | NHiBu | 3,4-Ph (2-Nph) | 1:1.1 | 1.81 ± 1.21 | 1.22 to 2.68 |
| 1 | 1.06 ± 1.21 | 0.72 to 1.55 | |||
| AMTB | 7.30 ± 1.50 | 7.85 to 8.82 |
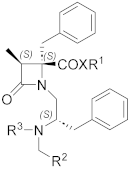 | |||||
| Compound | XR1 | R2 | R3 | IC50 (µM) | 95% Confidence Intervals |
|---|---|---|---|---|---|
| 1 | OtBu | 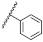 | Bn | 1.06 ± 1.21 | 0.72 to 1.55 |
| 34 | NHiBu | 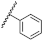 | Bn | 0.30 ± 1.26 | 0.19 to 0.50 |
| 35 | NHBn | 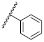 | Bn | 0.49 ± 1.27 | 0.30 to 0.79 |
| 36 | NHCH2(2-Pic) | 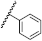 | Bn | 0.84 ± 1.32 | 0.48 to 1.49 |
| 37 | OtBu | 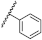 | H | 3.06 ± 1.25 | 2.67 to 3.51 |
| 40 | OtBu | 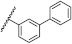 | H | 6.36 ± 1.41 | 3.08 to 13.17 |
| 41 | OtBu | 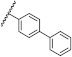 | H | 9.26 ± 1.41 | 4.44 to 19.35 |
| 42 | NHiBu | 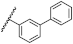 | H | 2.19 ± 1.36 | 1.15 to 4.17 |
| 43 | NHiBu | 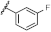 | H | 4.03 ± 1.35 | 2.14 to 7.60 |
| 44 | NHiBu | 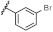 | H | 3.27 ± 1.31 | 1.86 to 5.75 |
| 45 | NHiBu | 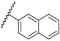 | H | 3.56 ± 1.40 | 1.74 to 7.31 |
| AMTB | 7.3 ± 1.50 | 6.82 to 7.85 | |||
| Compound | Ca2+ Microfluorimetry Assays | Patch-Clamp Assay | ||||
|---|---|---|---|---|---|---|
| rTRPM8 IC50 (µM) | 95% Confidence Intervals | hTRPM8 IC50 (µM) | 95% Confidence Intervals | rTRPM8 IC50 (µM) | 95% Confidence Intervals | |
| 1 | 1.06 ± 1.21 | 0.72 to 1.55 | 1.74 ± 1.19 | 1.23 to 2.45 | 0.60 ± 1.66 | 0.20 to 1.76 |
| 34 | 0.30 ± 1.26 | 0.19 to 0.50 | 2.58 ± 1.24 | 1.65 to 4.02 | 0.16 ± 0.82 | 0.10 to 0.271 |
| 35 | 0.49 ± 1.27 | 0.30 to 0.79 | 3.33 ± 1.28 | 2.03 to 5.48 | 0.48 ± 1.33 | 0.18 to 1.23 |
| Compound | % Channel Inhibition at 10 μM | % Inhibition of Radioligand Binding at 10 μM | ||||||
|---|---|---|---|---|---|---|---|---|
| hTRPV1 | hTRPV3 | hTRPA1 | hTRPM3 | ASIC3 | hCGRPR | hCB2 | hM3 | |
| 1 | 19.4 ± 1.7 | −6.3 ± 5.9 | 4.2 ± 1.1 | ND | 6.0 ± 0.9 | −1.0 ± 4.3 | 7.3 ± 0.3 | −4.1 ± 3.1 |
| 34 | 8.9 ± 2.9 | 17.3 ± 5.7 | 2.0 ± 3.4 | 6.9 ± 10.7 | 2.2 ± 9.6 | −11.9 ± 5.2 | 7.0 ± 2.1 | 5.2 ± 3.2 |
| Subsite | Channel Location | 34 | 35 | 37 | AMTB | Menthol |
|---|---|---|---|---|---|---|
| 1 | Pore, external tower | 20.0 (9.09) | 14.4 (7.82) | 28.2 (9.85) | 20 | 29.3 |
| 2 | Pore, high S3–S4, S6 | 13.8 (8.51) | 16.4 (10.11) | 7.4 (7.40) | 8.1 | 17.1 |
| 3 | Inner pore, S5S6, S5 loops | 29.3 (9.81) | 24.6 (9.40) | 31.8 (7.62) | 18.4 | 0 |
| 4 | Pore, internal mouth | 6.0 (11.21) | 7.6 (10.68) | 13.9 (8.49) | 18.6 | 0.3 |
| 5 | Menthol binding-site | 0 | 0 | 0 | 2.4 | 19.2 |
| 6 | S1–S4-TRP domain | 8.2 (9.28) | 10.6 (6.98) | 4.4 (6.27) | 10.9 | 18.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Escura, C.; Bonache, M.Á.; Medina, J.A.; Medina-Peris, A.; De Andrés-López, J.; González-Rodríguez, S.; Kerselaers, S.; Fernández-Ballester, G.; Voets, T.; Ferrer-Montiel, A.; et al. β-Lactam TRPM8 Antagonists Derived from Phe-Phenylalaninol Conjugates: Structure–Activity Relationships and Antiallodynic Activity. Int. J. Mol. Sci. 2023, 24, 14894. https://doi.org/10.3390/ijms241914894
Martín-Escura C, Bonache MÁ, Medina JA, Medina-Peris A, De Andrés-López J, González-Rodríguez S, Kerselaers S, Fernández-Ballester G, Voets T, Ferrer-Montiel A, et al. β-Lactam TRPM8 Antagonists Derived from Phe-Phenylalaninol Conjugates: Structure–Activity Relationships and Antiallodynic Activity. International Journal of Molecular Sciences. 2023; 24(19):14894. https://doi.org/10.3390/ijms241914894
Chicago/Turabian StyleMartín-Escura, Cristina, M. Ángeles Bonache, Jessy A. Medina, Alicia Medina-Peris, Jorge De Andrés-López, Sara González-Rodríguez, Sara Kerselaers, Gregorio Fernández-Ballester, Thomas Voets, Antonio Ferrer-Montiel, and et al. 2023. "β-Lactam TRPM8 Antagonists Derived from Phe-Phenylalaninol Conjugates: Structure–Activity Relationships and Antiallodynic Activity" International Journal of Molecular Sciences 24, no. 19: 14894. https://doi.org/10.3390/ijms241914894
APA StyleMartín-Escura, C., Bonache, M. Á., Medina, J. A., Medina-Peris, A., De Andrés-López, J., González-Rodríguez, S., Kerselaers, S., Fernández-Ballester, G., Voets, T., Ferrer-Montiel, A., Fernández-Carvajal, A., & González-Muñiz, R. (2023). β-Lactam TRPM8 Antagonists Derived from Phe-Phenylalaninol Conjugates: Structure–Activity Relationships and Antiallodynic Activity. International Journal of Molecular Sciences, 24(19), 14894. https://doi.org/10.3390/ijms241914894