
Chemical Inhibition of RPA by HAMNO Alters Cell Cycle Dynamics by Impeding DNA Replication and G2-to-M Transition but Has Little Effect on the Radiation-Induced DNA Damage Response
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
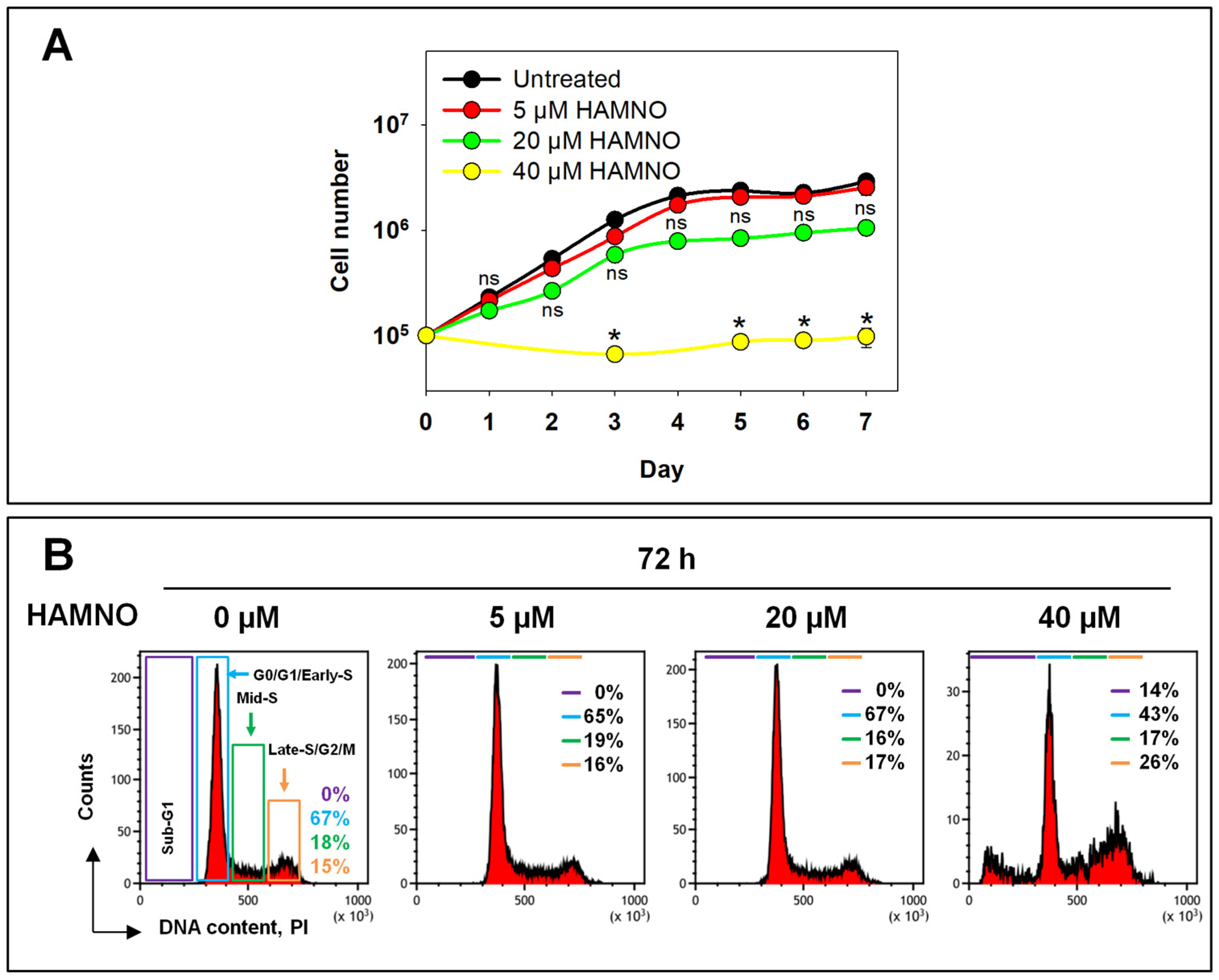
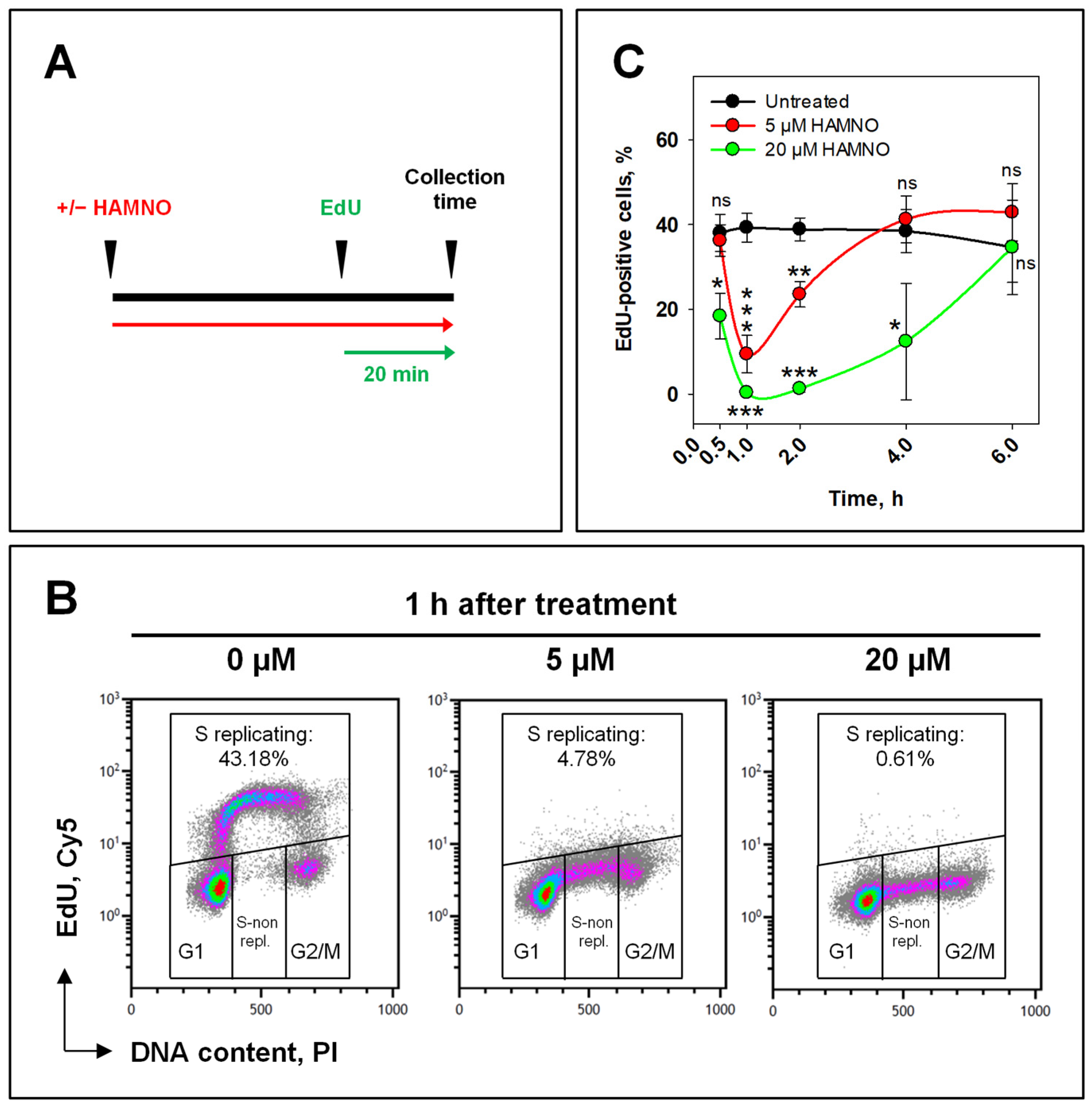
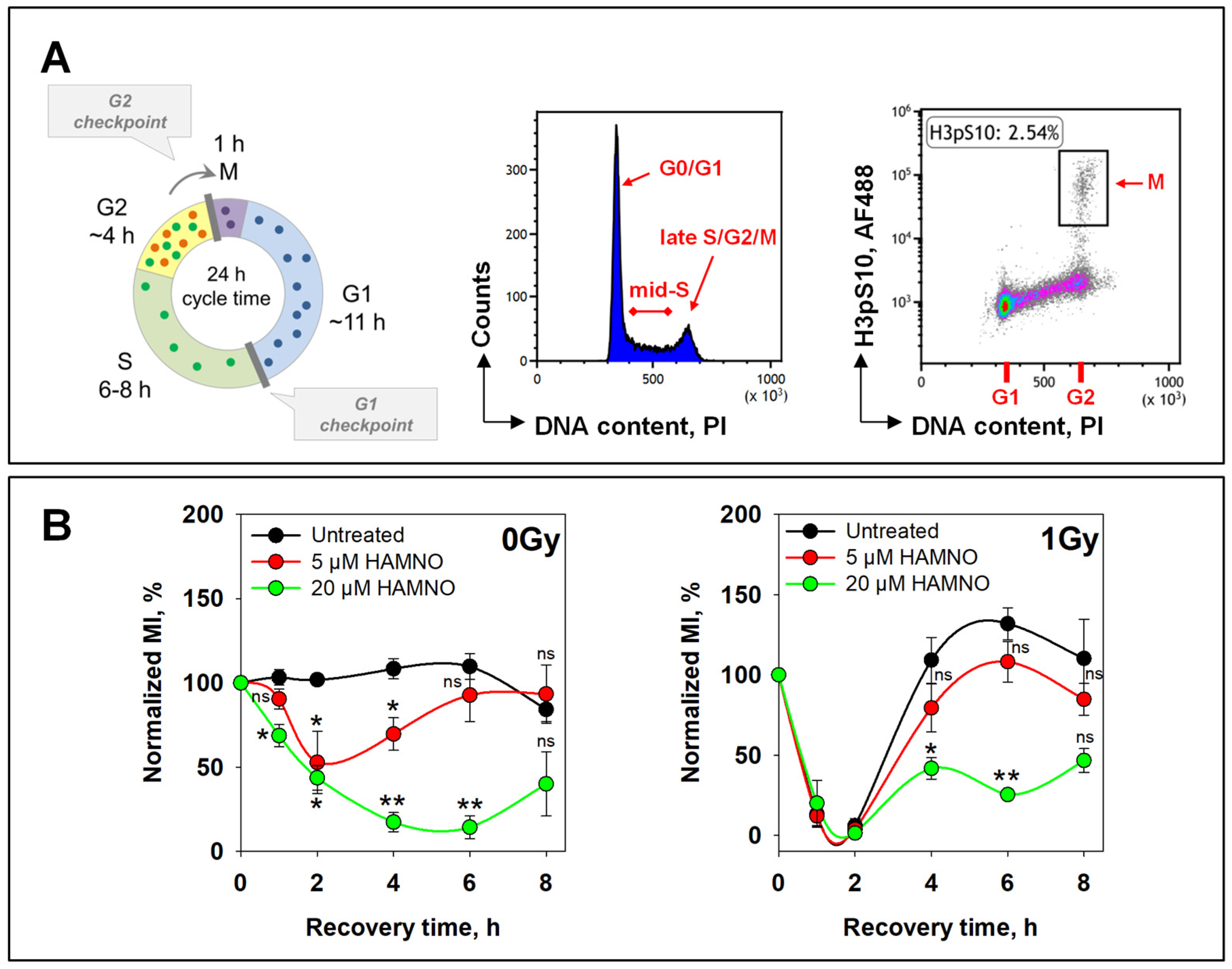
2.1. RPA Inhibition by HAMNO Transiently Inhibits Cell Growth and DNA Synthesis
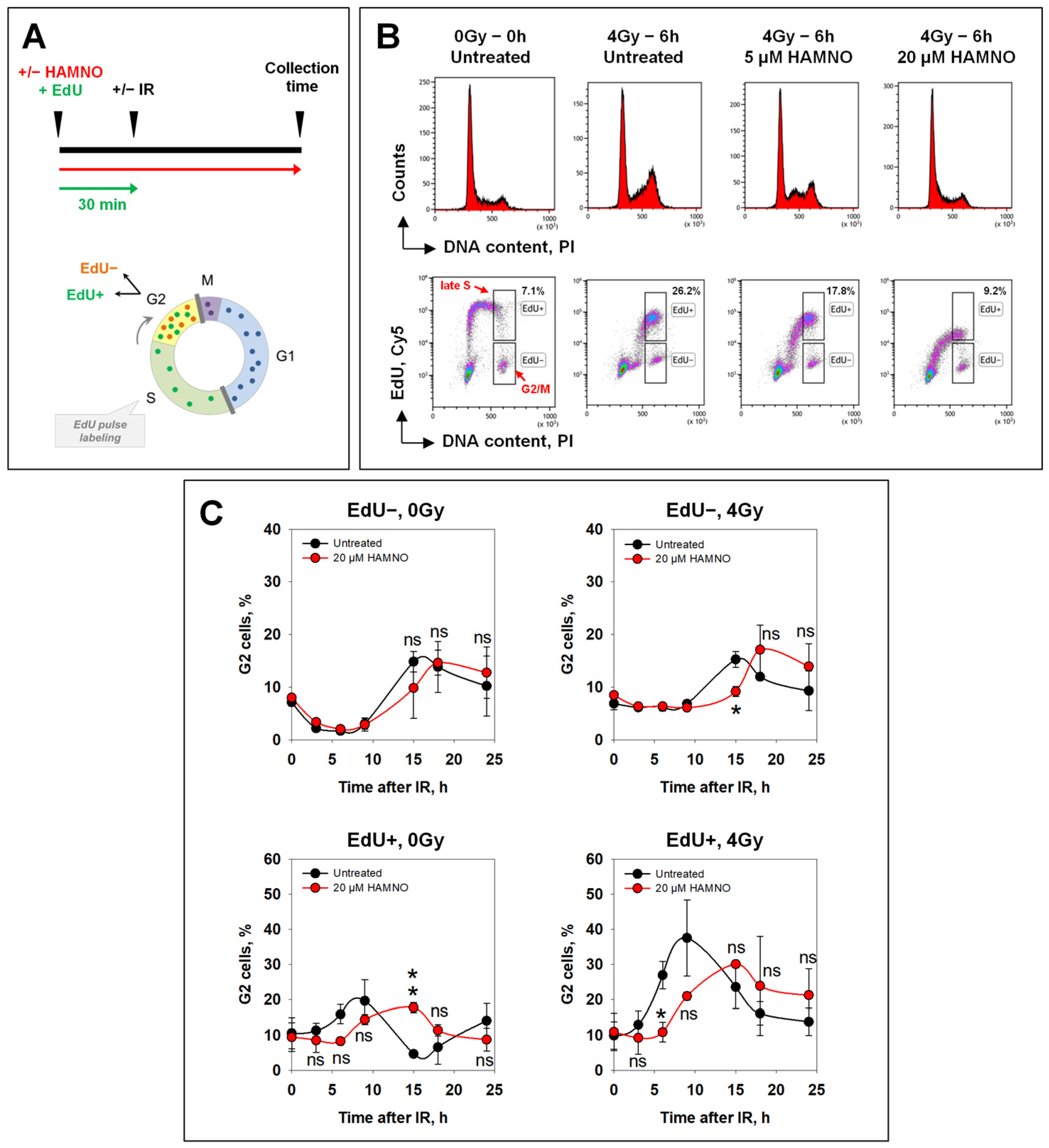
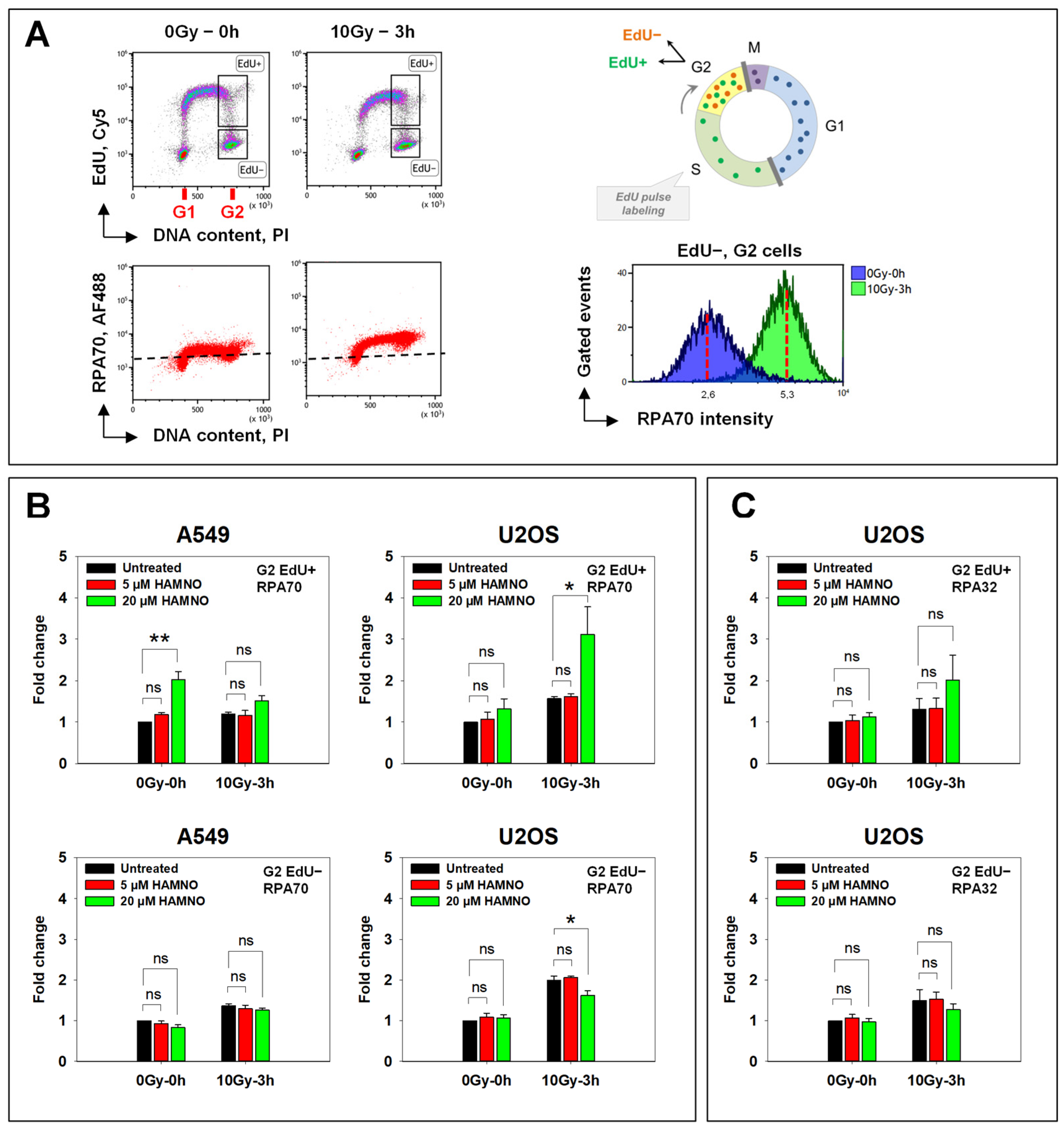
2.2. HAMNO Enhances IR-Related Resection in S-Phase Irradiated Cells in a Cell Line-Specific Manner
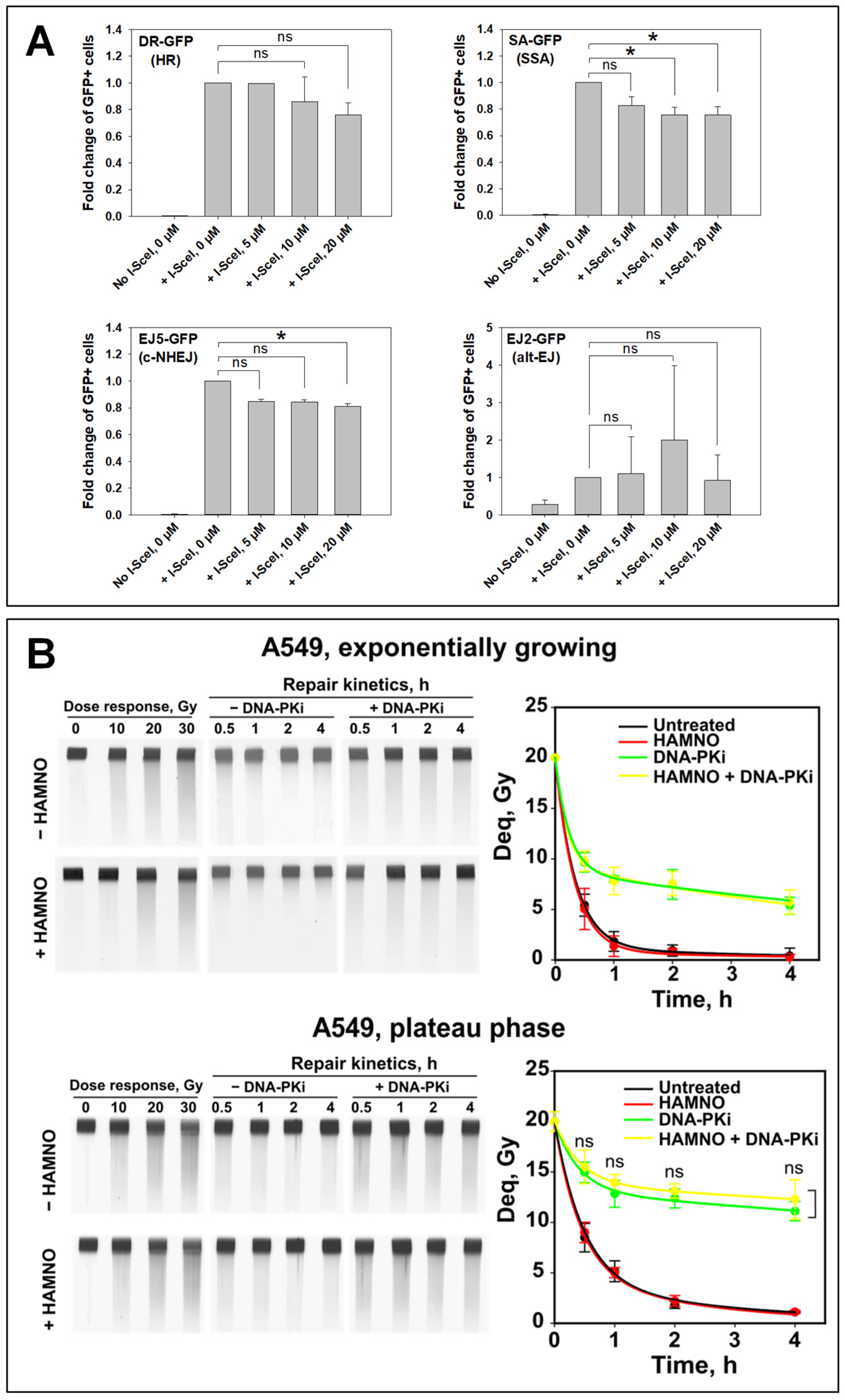
2.3. HAMNO Abrogates Repair by HR in a Cell Type-Specific Manner
2.4. HAMNO Fails to Radiosensitize A549 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Growth
4.2. Treatment with IR and Small Molecule Inhibitors
4.3. Flow Cytometry Analysis of DNA Replication by EdU Incorporation
4.4. Quantification of Mitotic Index by Flow Cytometry
4.5. Flow Cytometry Analysis of Chromatin-Bound RPA
4.6. I-SceI-Based Reporter Assays for Assessing DSB Repair by Different Repair Pathways
4.7. Immunofluorescence and Quantitative Image-Based Cytometry (QIBC)
4.8. Clonogenic Survival Assay
4.9. Pulse-Field Gel Electrophoresis (PFGE)
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Replication protein A: A multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. End Resection at Double-Strand Breaks: Mechanism and Regulation. Cold Spring Harb. Perspect. Biol. 2014, 6, a016436. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder. BioEssays 2014, 36, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef] [PubMed]
- Soniat, M.M.; Myler, L.R.; Kuo, H.-C.; Paull, T.T.; Finkelstein, I.J. RPA Phosphorylation Inhibits DNA Resection. Mol. Cell 2019, 75, 145–153.e145. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Stillman, B. cdc2 family kinases phosphorylate a human cell DNA replication factor, RPA, and activate DNA replication. EMBO J. 1992, 11, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Newport, J.W. Distinct roles of cdk2 and cdc2 in RP-A phosphorylation during the cell cycle. J. Cell Sci. 1993, 106, 983–994. [Google Scholar] [CrossRef]
- Niu, H.; Erdjument-Bromage, H.; Pan, Z.-Q.; Lee, S.-H.; Tempst, P.; Hurwitz, J. Mapping of amino acid residues in the p34 subunit of human single-stranded DNA-binding protein phosphorylated by DNA-dependent protein kinase and Cdc2 kinase in Vitro. J. Biol. Chem. 1997, 272, 12634–12641. [Google Scholar] [CrossRef]
- Zernik-Kobak, M.; Vasunia, K.; Connelly, M.; Anderson, C.W. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J. Biol. Chem. 1997, 272, 23896–23904. [Google Scholar] [CrossRef]
- Shao, R.-G.; Cao, C.-X.; Zhang, H.; Kohn, K.W.; Wold, M.S.; Pommier, Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999, 18, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guan, J.; Wang, H.; Perrault, A.R.; Wang, Y.; Iliakis, G. Replication Protein A2 Phosphorylation after DNA Damage by the Coordinated Action of Ataxia Telangiectasia-Mutated and DNA-dependent Protein Kinase. Cancer Res. 2001, 61, 8554–8563. [Google Scholar] [PubMed]
- Sakasai, R.; Shinohe, K.; Ichijima, Y.; Okita, N.; Shibata, A.; Asahina, K.; Teraoka, H. Differential involvement of phosphatidylinositol 3-kinase-related protein kinases in hyperphosphorylation of replication protein A2 in response to replication-mediated DNA double-strand breaks. Genes Cells 2006, 11, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Cruet-Hennequart, S.; Coyne, S.; Glynn, M.T.; Oakley, G.G.; Carty, M.P. UV-induced RPA phosphorylation is increased in the absence of DNA polymerase [eta] and requires DNA-PK. DNA Repair 2006, 5, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.; Nievera, C.J.; Klimovich, V.; Fanning, E.; Wu, X. RPA2 Is a Direct Downstream Target for ATR to Regulate the S-phase Checkpoint. J. Biol. Chem. 2006, 281, 39517–39533. [Google Scholar] [CrossRef]
- Vassin, V.M.; Anantha, R.W.; Sokolova, E.; Kanner, S.; Borowiec, J.A. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J. Cell Sci. 2009, 122, 4070–4080. [Google Scholar] [CrossRef] [PubMed]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Block, W.D.; Yu, Y.; Lees-Miller, S.P. Phosphatidyl inositol 3-kinase-like serine/threonine protein kinases (PIKKs) are required for DNA damage-induced phosphorylation of the 32 kDa subunit of replication protein A at threonine 21. Nucleic Acids Res. 2004, 32, 997–1005. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.; Liu, Y.; Zou, Y. Preferential localization of hyperphosphorylated replication protein A to double-strand break repair and checkpoint complexes upton DNA damage. Biochem. J. 2005, 391, 473–480. [Google Scholar] [CrossRef]
- Forment, J.V.; Walker, R.V.; Jackson, S.P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytom. Part A 2012, 81A, 922–928. [Google Scholar] [CrossRef]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G2-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2020, 48, 1905–1924. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mladenov, E.; Mortoga, S.; Iliakis, G. SCF(SKP2) regulates APC/C(CDH1)-mediated degradation of CTIP to adjust DNA-end resection in G2-phase. Cell Death Dis. 2020, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Fowler, F.C.; Chen, B.-R.; Zolnerowich, N.; Wu, W.; Pavani, R.; Paiano, J.; Peart, C.; Nussenzweig, A.; Sleckman, B.P.; Tyler, J.K. DNA-PK Promotes DNA End Resection at DNA Double Strand Breaks in G0 cells. bioRxiv 2021, 11, e74700. [Google Scholar] [CrossRef]
- Li, F.; Mladenov, E.; Dueva, R.; Stuschke, M.; Timmermann, B.; Iliakis, G. Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation. Cells 2022, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.R.; Tyler, J.K.; Sleckman, B.P. A Flow Cytometry-Based Method for Analyzing DNA End Resection in G0- and G1-Phase Mammalian Cells. Bio-Protoc. 2022, 12, e4413. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Liu, S.; Wang, L.; Mosel, A.; Peng, A.; Oakley, G.G. RPA Inhibition Increases Replication Stress and Suppresses Tumor Growth. Cancer Res. 2014, 74, 5165–5172. [Google Scholar] [CrossRef]
- Shuck, S.C.; Turchi, J.J. Targeted Inhibition of Replication Protein A Reveals Cytotoxic Activity, Synergy with Chemotherapeutic DNA-Damaging Agents, and Insight into Cellular Function. Cancer Res. 2010, 70, 3189–3198. [Google Scholar] [CrossRef]
- Glanzer, J.G.; Liu, S.; Oakley, G.G. Small molecule inhibitor of the RPA70 N-terminal protein interaction domain discovered using in silico and in vitro methods. Bioorganic Med. Chem. 2011, 19, 2589–2595. [Google Scholar] [CrossRef]
- Glanzer, J.G.; Carnes, K.A.; Soto, P.; Liu, S.; Parkhurst, L.J.; Oakley, G.G. A small molecule directly inhibits the p53 transactivation domain from binding to replication protein A. Nucleic Acids Res. 2013, 41, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Bochkareva, E.; Kaustov, L.; Ayed, A.; Yi, G.S.; Lu, Y.; Pineda-Lucena, A.; Liao, J.C.; Okorokov, A.L.; Milner, J.; Arrowsmith, C.H.; et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. USA 2005, 102, 15412–15417. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Vaithiyalingam, S.; Glick, G.G.; Mordes, D.A.; Chazin, W.J.; Cortez, D. The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol. Cell. Biol. 2008, 28, 7345–7353. [Google Scholar] [CrossRef] [PubMed]
- Oakley, G.G.; Tillison, K.; Opiyo, S.; Glanzer, J.; Horn, J.M.; Patrick, S.M. Physical Interaction between Replication Protein A (RPA) and MRN: Involvement of RPA2 Phosphorylation and the N-terminus of RPA1. Biochemistry 2009, 48, 7473–7481. [Google Scholar] [CrossRef] [PubMed]
- Guilliam, T.A.; Jozwiakowski, S.K.; Ehlinger, A.; Barnes, R.P.; Rudd, S.G.; Bailey, L.J.; Skehel, J.M.; Eckert, K.A.; Chazin, W.J.; Doherty, A.J. Human PrimPol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res. 2015, 43, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Zhao, Y.; Xu, Y.; Ning, S.; Hou, W.; Hou, M.; Gao, G.; Ji, J.; Guo, R.; Xu, D. Ewing Tumor-Associated Antigen 1 Interacts with Replication Protein A to Promote Restart of Stalled Replication Forks. J. Biol. Chem. 2016, 291, 21956–21962. [Google Scholar] [CrossRef] [PubMed]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Raschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef]
- Lee, Y.C.; Zhou, Q.; Chen, J.; Yuan, J. RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr. Biol. 2016, 26, 3257–3268. [Google Scholar] [CrossRef]
- Guilliam, T.A.; Brissett, N.C.; Ehlinger, A.; Keen, B.A.; Kolesar, P.; Taylor, E.M.; Bailey, L.J.; Lindsay, H.D.; Chazin, W.J.; Doherty, A.J. Molecular basis for PrimPol recruitment to replication forks by RPA. Nat. Commun. 2017, 8, 15222. [Google Scholar] [CrossRef] [PubMed]
- Binz, S.K.; Lao, Y.; Lowry, D.F.; Wold, M.S. The phosphorylation domain of the 32-kDa subunit of replication protein A (RPA) modulates RPA-DNA interactions. Evidence for an intersubunit interaction. J. Biol. Chem. 2003, 278, 35584–35591. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in Checkpoint Signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Kim, S.-t.; Kastan, M.B. Involvement of Brca1 in S-Phase and G2-Phase Checkpoints after Ionizing Irradiation. Mol. Cell. Biol. 2001, 21, 3445–3450. [Google Scholar] [CrossRef]
- Xu, B.; Kim, S.T.; Lim, D.S.; Kastan, M.B. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol. Cell. Biol. 2002, 22, 1049–1059. [Google Scholar] [CrossRef]
- Shibata, A.; Barton, O.; Noon, A.T.; Dahm, K.; Deckbar, D.; Goodarzi, A.A.; Lobrich, M.; Jeggo, P.A. Role of ATM and the Damage Response Mediator Proteins 53BP1 and MDC1 in the Maintenance of G2/M Checkpoint Arrest. Mol. Cell. Biol. 2010, 30, 3371–3383. [Google Scholar] [CrossRef]
- Sung, P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 1994, 265, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef]
- Sugiyama, T.; Zaitseva, E.M.; Kowalczykowski, S.C. A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the saccharomyces cerevisiae Rad51 protein. J. Biol. Chem. 1997, 272, 7940–7945. [Google Scholar] [CrossRef]
- Golub, E.I.; Gupta, R.C.; Haaf, T.; Wold, M.S.; Radding, C.M. Interaction of human Rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res. 1998, 26, 5388–5393. [Google Scholar] [CrossRef]
- Song, B.-W.; Sung, P. Functional Interactions among Yeast Rad51 Recombinase, Rad52 Mediator, and Replication Protein A in DNA Strand Exchange. J. Biol. Chem. 2000, 275, 15895–15904. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kowalczykowski, S.C. Rad52 Protein Associates with Replication Protein A (RPA)-Single-stranded DNA to Accelerate Rad51-mediated Displacement of RPA and Presynaptic Complex Formation. J. Biol. Chem. 2002, 277, 31663–31672. [Google Scholar] [CrossRef] [PubMed]
- Kantake, N.; Sugiyama, T.; Kolodner, R.D.; Kowalczykowski, S.C. The Recombination-deficient Mutant RPA (rfat-t11) Is Displaced Slowly from Single-stranded DNA by Rad51 Protein. J. Biol. Chem. 2003, 278, 23410–23417. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, M.E.; Chazin, W.J. Physical Interaction between Replication Protein A and Rad51 Promotes Exchange on Single-stranded DNA. J. Biol. Chem. 2004, 279, 25638–25645. [Google Scholar] [CrossRef]
- Wang, X.; Haber, J.E. Role of Saccharomyces Single-Stranded DNA-Binding Protein RPA in the Strand Invasion Step of Double-Strand Break Repair. PLoS Biol. 2004, 2, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.-B.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.; Stark, J.M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 2012, 920, 379–391. [Google Scholar] [CrossRef]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef]
- Windhofer, F.; Wu, W.; Wang, M.; Singh, S.K.; Saha, J.; Rosidi, B.; Iliakis, G. Marked dependence on growth state of backup pathways of NHEJ. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 1462–1470. [Google Scholar] [CrossRef]
- Singh, S.K.; Wu, W.; Zhang, L.; Klammer, H.; Wang, M.; Iliakis, G. Widespread Dependence of Backup NHEJ on Growth State: Ramifications for the Use of DNA-PK Inhibitors. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 540–548. [Google Scholar] [CrossRef]
- Sinclair, W.K. Cyclic x-ray responses in mammalian cells in vitro. Radiat. Res. 1968, 33, 620–643. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Nusse, M. Aphidicolin promotes repair of potentially lethal damage in irradiated mammalian cells synchronized in S-phase. Biochem. Biophys. Res. Commun. 1982, 4, 1209–1214. [Google Scholar] [CrossRef]
- Nguyen, B.; Sokoloski, J.; Galletto, R.; Elson, E.L.; Wold, M.S.; Lohman, T.M. Diffusion of Human Replication Protein A along Single-Stranded DNA. J. Mol. Biol. 2014, 426, 3246–3261. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Subramanyam, S.; Elcock, A.H.; Spies, M.; Wold, M.S. Dynamic binding of replication protein a is required for DNA repair. Nucleic Acids Res. 2016, 44, 5758–5772. [Google Scholar] [CrossRef]
- Mishra, G.; Bigman, L.S.; Levy, Y. ssDNA diffuses along replication protein A via a reptation mechanism. Nucleic Acids Res. 2020, 48, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Spegg, V.; Panagopoulos, A.; Stout, M.; Krishnan, A.; Reginato, G.; Imhof, R.; Roschitzki, B.; Cejka, P.; Altmeyer, M. Phase separation properties of RPA combine high-affinity ssDNA binding with dynamic condensate functions at telomeres. Nat. Struct. Mol. Biol. 2023, 30, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Magin, S.; Papaioannou, M.; Saha, J.; Staudt, C.; Iliakis, G.E. Inhibition of homologous recombination and promotion of mutagenic repair of DNA double-strand breaks underpins arabinoside-nucleoside analog-radiosensitization. Mol. Cancer Ther. 2015, 14, 1424–1433. [Google Scholar] [CrossRef]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell. 2017, 65, 336–346. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell. Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Pedersen, H.; Anne Adanma Obara, E.; Elbaek, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. Int. J. Mol. Sci. 2020, 21, 1588. [Google Scholar] [CrossRef]
- Abramova, N.A.; Russell, J.; Botchan, M.; Li, R. Interaction between replication protein A and p53 is disrupted after UV damage in a DNA repair-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 7186–7191. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.A.; Li, Z.; Dangeti, M.; Musich, P.R.; Patrick, S.; Roginskaya, M.; Cartwright, B.; Zou, Y. DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair. Oncogene 2013, 32, 2452–2462. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Kim, J.M. The RPA inhibitor HAMNO sensitizes Fanconi anemia pathway-deficient cells. Cell Cycle 2022, 21, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.N.; Engelberg, J. Hela Cells: Effects of Temperature on the Life Cycle. Science 1965, 148, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Kenny, M.K.; Schlegel, U.; Furneaux, H.; Hurwitz, J. The role of human single-stranded DNA binding protein and its individual subunits in simian virus 40 DNA replication. J. Biol. Chem. 1990, 265, 7693–7700. [Google Scholar] [CrossRef] [PubMed]
- Metzger, L.; Iliakis, G. Kinetics of DNA double strand breaks throughout the cell cycle as assayed by pulsed field gel electrophoresis in CHO cells. Int. J. Radiat. Biol. 1991, 59, 1325–1339. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, M.; Wu, W.; Singh, S.K.; Mussfeldt, T.; Iliakis, G. Repair of radiation induced DNA double strand breaks by backup NHEJ is enhanced in G2. DNA Repair 2008, 7, 329–338. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dueva, R.; Krieger, L.M.; Li, F.; Luo, D.; Xiao, H.; Stuschke, M.; Metzen, E.; Iliakis, G. Chemical Inhibition of RPA by HAMNO Alters Cell Cycle Dynamics by Impeding DNA Replication and G2-to-M Transition but Has Little Effect on the Radiation-Induced DNA Damage Response. Int. J. Mol. Sci. 2023, 24, 14941. https://doi.org/10.3390/ijms241914941
Dueva R, Krieger LM, Li F, Luo D, Xiao H, Stuschke M, Metzen E, Iliakis G. Chemical Inhibition of RPA by HAMNO Alters Cell Cycle Dynamics by Impeding DNA Replication and G2-to-M Transition but Has Little Effect on the Radiation-Induced DNA Damage Response. International Journal of Molecular Sciences. 2023; 24(19):14941. https://doi.org/10.3390/ijms241914941
Chicago/Turabian StyleDueva, Rositsa, Lisa Marie Krieger, Fanghua Li, Daxian Luo, Huaping Xiao, Martin Stuschke, Eric Metzen, and George Iliakis. 2023. "Chemical Inhibition of RPA by HAMNO Alters Cell Cycle Dynamics by Impeding DNA Replication and G2-to-M Transition but Has Little Effect on the Radiation-Induced DNA Damage Response" International Journal of Molecular Sciences 24, no. 19: 14941. https://doi.org/10.3390/ijms241914941