
AZD-7648, a DNA-PK Inhibitor, Induces DNA Damage, Apoptosis, and Cell Cycle Arrest in Chronic and Acute Myeloid Leukemia Cells

 , , ,
, , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. DNA Damage Response Characterization of CML and AML Cell Lines
2.1.1. Mutational Status of DDR Genes
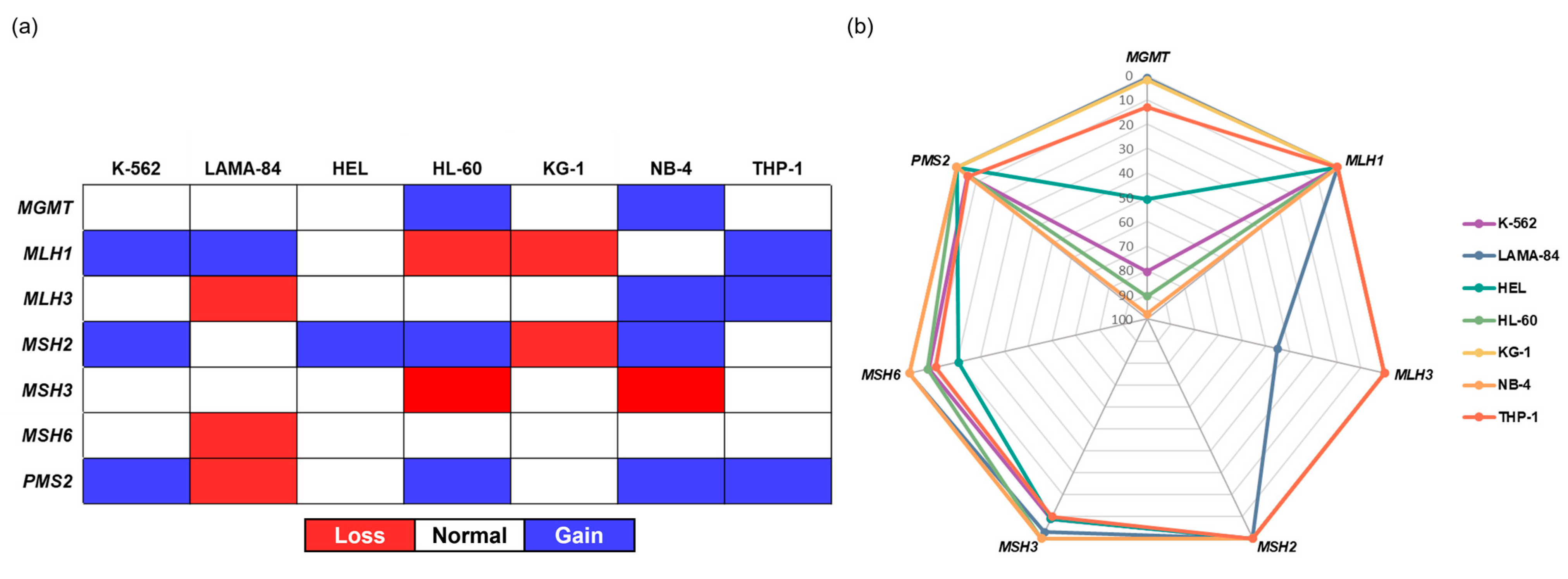
2.1.2. Genetic and Epigenetic Characterization of Selected DDR Genes
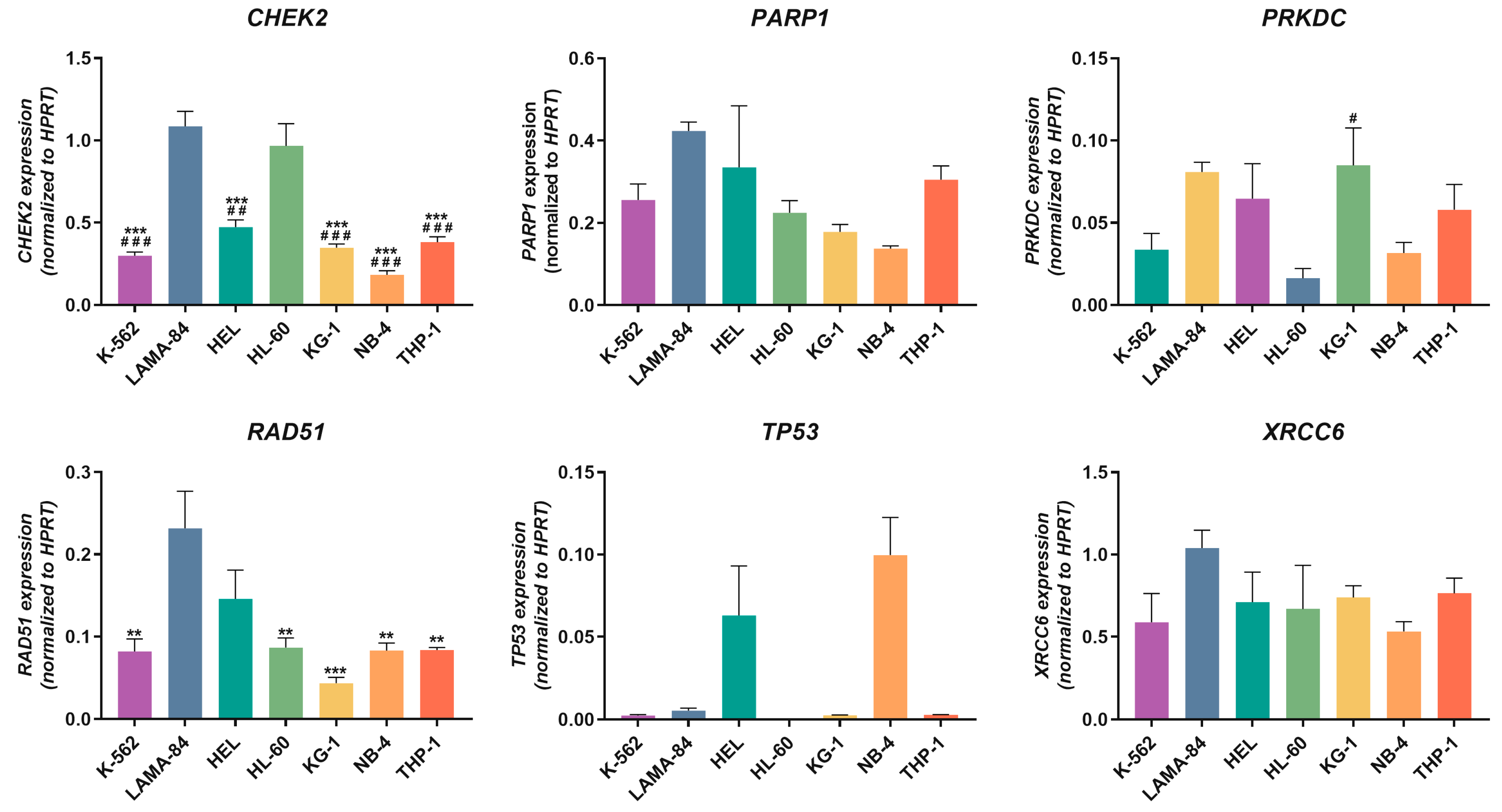
2.1.3. DDR-Related Genes’ Expression Levels
2.1.4. p53 Protein Expression
2.1.5. DNA and Chromosomal Damage Levels
2.1.6. Double-Strand Break Repair Kinetics
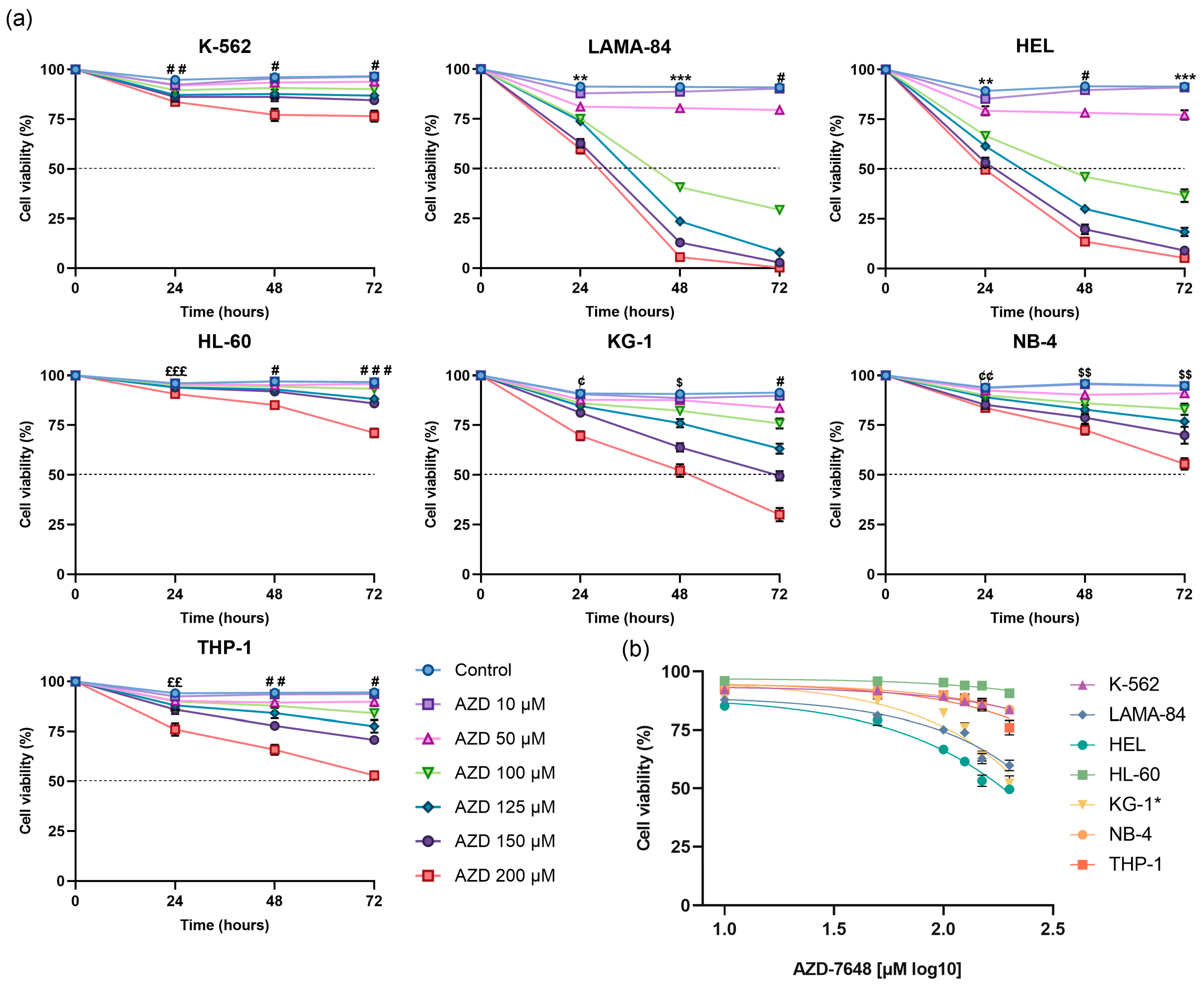
2.2. AZD-7648 Reduced Cell Density and Viability in AML Cell Lines
2.3. Apoptosis was the Main Mechanism of Cell Death Induced by AZD-7648
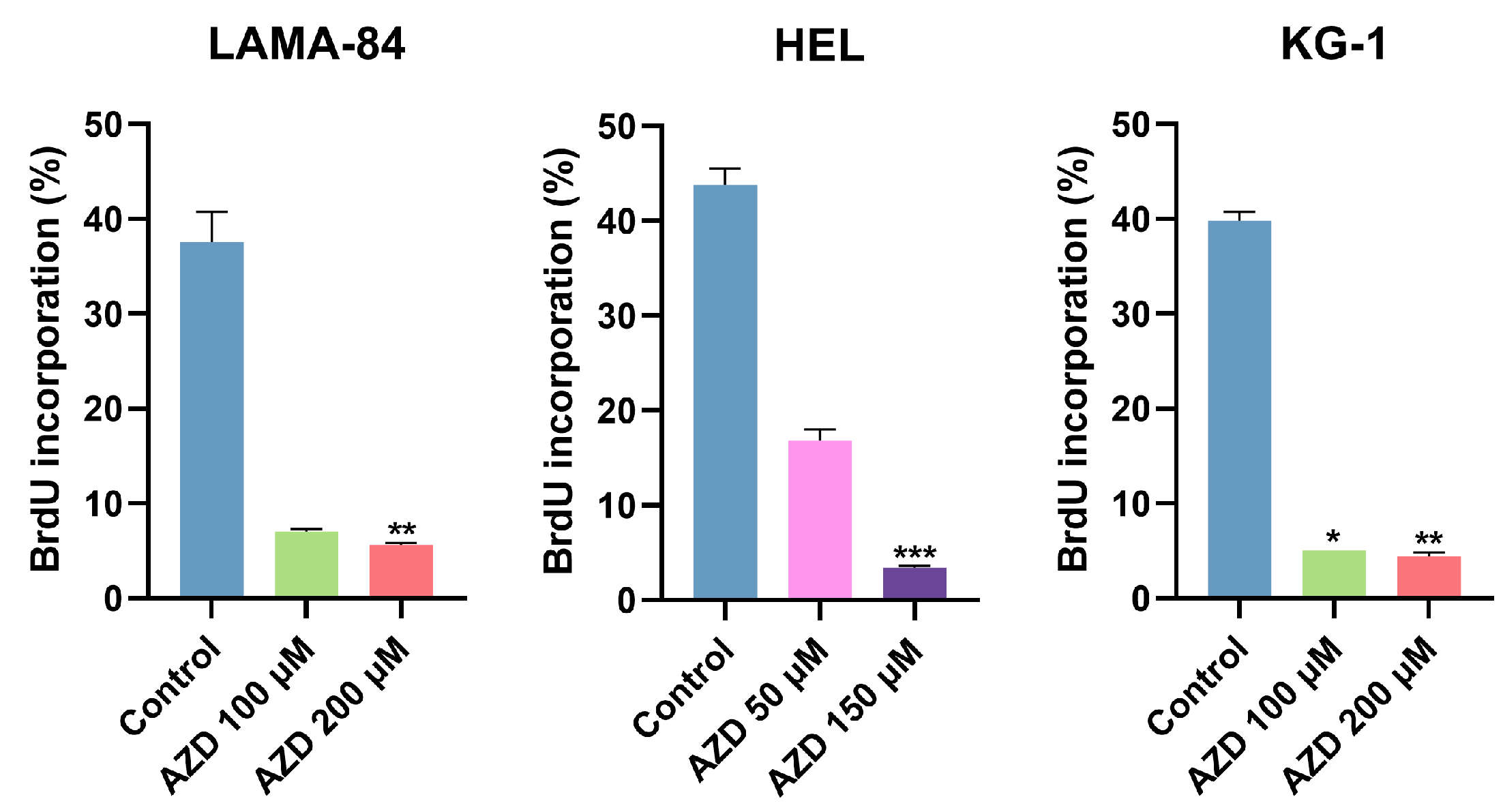
2.4. AZD-7648 Promotes Cell Cycle Arrest in G0/G1 Phase and Reduces Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. DDR Pathways Mutations Database Analysis
4.3. Methylation-Specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA)
4.4. Gene Expression Analysis
4.5. p53 Expression
4.6. γH2AX Expression Assessment
4.7. Cytokinesis-Block Micronucleus Cytome Assay (CBMN)
4.8. Telomere Length Quantification
4.9. Double-Strand Breaks Repair Evaluation
4.10. Cell Density and Viability Analysis
4.11. Cell Death Analysis
4.12. Cell Cycle Analysis
4.13. Apoptosis, DNA Damage, and Cell Proliferation Assessment
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C. The DNA damage response—From cell biology to human disease. J. Transl. Genet. Genom. 2022, 6, 204–222. [Google Scholar] [CrossRef]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Knudsen, K.E. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef]
- Wang, C.; Tang, M.; Chen, Z.; Nie, L.; Li, S.; Xiong, Y.; Szymonowicz, K.A.; Park, J.M.; Zhang, H.; Feng, X.; et al. Genetic vulnerabilities upon inhibition of DNA damage response. Nucleic Acids Res. 2021, 49, 8214–8231. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Nilles, N.; Fahrenkrog, B. Taking a Bad Turn: Compromised DNA Damage Response in Leukemia. Cells 2017, 6, 11. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Mustafa, A.G.; Malki, M.I. Targeting DNA Repair Pathways in Hematological Malignancies. Int. J. Mol. Sci. 2020, 21, 7365. [Google Scholar] [CrossRef] [PubMed]
- Popp, H.D.; Kohl, V.; Naumann, N.; Flach, J.; Brendel, S.; Kleiner, H.; Weiss, C.; Seifarth, W.; Saussele, S.; Hofmann, W.K.; et al. DNA Damage and DNA Damage Response in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 1177. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Goncalves, A.C.; Rutella, S.; Almeida, A.M.; De Las Rivas, J.; Trougakos, I.P.; Sarmento Ribeiro, A.B. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia-From Molecular Mechanisms to Clinical Relevance. Cancers 2021, 13, 4820. [Google Scholar] [CrossRef] [PubMed]
- Horibata, S.; Alyateem, G.; DeStefano, C.B.; Gottesman, M.M. The Evolving AML Genomic Landscape: Therapeutic Implications. Curr. Cancer Drug Targets 2020, 20, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Trino, S.; Laurenzana, I.; Lamorte, D.; Calice, G.; De Stradis, A.; Santodirocco, M.; Sgambato, A.; Caivano, A.; De Luca, L. Acute Myeloid Leukemia Cells Functionally Compromise Hematopoietic Stem/Progenitor Cells Inhibiting Normal Hematopoiesis Through the Release of Extracellular Vesicles. Front. Oncol. 2022, 12, 824562. [Google Scholar] [CrossRef]
- Esposito, M.T.; So, C.W. DNA damage accumulation and repair defects in acute myeloid leukemia: Implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014, 123, 545–561. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Mufti, G.J.; Rassool, F.V. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002, 62, 2791–2797. [Google Scholar]
- Dylgjeri, E.; Knudsen, K.E. DNA-PKcs: A Targetable Protumorigenic Protein Kinase. Cancer Res. 2022, 82, 523–533. [Google Scholar] [CrossRef]
- Damia, G. Targeting DNA-PK in cancer. Mutat. Res. 2020, 821, 111692. [Google Scholar] [CrossRef]
- Kelm, J.M.; Samarbakhsh, A.; Pillai, A.; VanderVere-Carozza, P.S.; Aruri, H.; Pandey, D.S.; Pawelczak, K.S.; Turchi, J.J.; Gavande, N.S. Recent Advances in the Development of Non-PIKKs Targeting Small Molecule Inhibitors of DNA Double-Strand Break Repair. Front. Oncol. 2022, 12, 850883. [Google Scholar] [CrossRef]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Hui, Z.; Lirussi, F.; Garrido, C.; Ye, X.Y.; Xie, T. Small molecule DNA-PK inhibitors as potential cancer therapy: A patent review (2010-present). Expert Opin. Ther. Pat. 2021, 31, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Derman, B.A.; Larson, R.A. Post-remission therapy in acute myeloid leukemia: Are we ready for an individualized approach? Best Pract. Res. Clin. Haematol. 2019, 32, 101102. [Google Scholar] [CrossRef]
- Hafsi, H.; Dillon, M.T.; Barker, H.E.; Kyula, J.N.; Schick, U.; Paget, J.T.; Smith, H.G.; Pedersen, M.; McLaughlin, M.; Harrington, K.J. Combined ATR and DNA-PK Inhibition Radiosensitizes Tumor Cells Independently of Their p53 Status. Front. Oncol. 2018, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Firsanov, D.V.; Solovjeva, L.V.; Svetlova, M.P. H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clin. Epigenetics 2011, 2, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 2010, 920161. [Google Scholar] [CrossRef]
- Banath, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual gammaH2AX foci as an indication of lethal DNA lesions. BMC Cancer 2010, 10, 4. [Google Scholar] [CrossRef]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2020, 11, 629266. [Google Scholar] [CrossRef]
- Madhusudan, S.; Middleton, M.R. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat. Rev. 2005, 31, 603–617. [Google Scholar] [CrossRef]
- Elbakry, A.; Lobrich, M. Homologous Recombination Subpathways: A Tangle to Resolve. Front Genet 2021, 12, 723847. [Google Scholar] [CrossRef]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Obe, G.; Durante, M. DNA double strand breaks and chromosomal aberrations. Cytogenet. Genome Res. 2010, 128, 8–16. [Google Scholar] [CrossRef]
- Luzhna, L.; Kathiria, P.; Kovalchuk, O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front. Genet. 2013, 4, 131. [Google Scholar] [CrossRef] [PubMed]
- Takeiri, A.; Matsuzaki, K.; Motoyama, S.; Yano, M.; Harada, A.; Katoh, C.; Tanaka, K.; Mishima, M. High-content imaging analyses of gammaH2AX-foci and micronuclei in TK6 cells elucidated genotoxicity of chemicals and their clastogenic/aneugenic mode of action. Genes Environ. 2019, 41, 4. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007, 2, 1084–1104. [Google Scholar] [CrossRef] [PubMed]
- Coluzzi, E.; Colamartino, M.; Cozzi, R.; Leone, S.; Meneghini, C.; O’Callaghan, N.; Sgura, A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE 2014, 9, e110963. [Google Scholar] [CrossRef]
- Jdey, W.; Thierry, S.; Popova, T.; Stern, M.H.; Dutreix, M. Micronuclei Frequency in Tumors Is a Predictive Biomarker for Genetic Instability and Sensitivity to the DNA Repair Inhibitor AsiDNA. Cancer Res. 2017, 77, 4207–4216. [Google Scholar] [CrossRef]
- Cai, M.Y.; Dunn, C.E.; Chen, W.; Kochupurakkal, B.S.; Nguyen, H.; Moreau, L.A.; Shapiro, G.I.; Parmar, K.; Kozono, D.; D’Andrea, A.D. Cooperation of the ATM and Fanconi Anemia/BRCA Pathways in Double-Strand Break End Resection. Cell Rep. 2020, 30, 2402–2415.e2405. [Google Scholar] [CrossRef]
- Eccles, L.J.; Bell, A.C.; Powell, S.N. Inhibition of non-homologous end joining in Fanconi Anemia cells results in rescue of survival after interstrand crosslinks but sensitization to replication associated double-strand breaks. DNA Repair 2018, 64, 1–9. [Google Scholar] [CrossRef]
- Hanawalt, P.C. Historical perspective on the DNA damage response. DNA Repair 2015, 36, 2–7. [Google Scholar] [CrossRef]
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Myung, K. Crosstalk between different DNA repair pathways for DNA double strand break repairs. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2022, 873, 503438. [Google Scholar] [CrossRef] [PubMed]
- Spies, M.; Fishel, R. Mismatch repair during homologous and homeologous recombination. Cold Spring Harb. Perspect. Biol. 2015, 7, a022657. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.J.; Chao, H.X.; Roberts, V.R.; Sullivan, A.R.; Shah, S.J.; Simpson, D.A.; Feng, W.; Wozny, A.-S.; Kumar, S.; Purvis, J.E.; et al. Hyperactive end joining repair mediates resistance to DNA damaging therapy in p53-deficient cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kumar, R.J.; Chao, H.X.; Simpson, D.A.; Feng, W.; Cho, M.G.; Roberts, V.R.; Sullivan, A.R.; Shah, S.J.; Wozny, A.S.; Fagan-Solis, K.; et al. Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency. NAR Cancer 2020, 2, zcaa038. [Google Scholar] [CrossRef]
- Wolf, D.; Rotter, V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc. Natl. Acad. Sci. USA 1985, 82, 790–794. [Google Scholar] [CrossRef]
- Murad, H.; Hawat, M.; Ekhtiar, A.; AlJapawe, A.; Abbas, A.; Darwish, H.; Sbenati, O.; Ghannam, A. Induction of G1-phase cell cycle arrest and apoptosis pathway in MDA-MB-231 human breast cancer cells by sulfated polysaccharide extracted from Laurencia papillosa. Cancer Cell Int. 2016, 16, 39. [Google Scholar] [CrossRef]
- Yan, Y.Q.; Zhang, B.; Wang, L.; Xie, Y.H.; Peng, T.; Bai, B.; Zhou, P.K. Induction of apoptosis and autophagic cell death by the vanillin derivative 6-bromine-5-hydroxy-4-methoxybenzaldehyde is accompanied by the cleavage of DNA-PKcs and rapid destruction of c-Myc oncoprotein in HepG2 cells. Cancer Lett. 2007, 252, 280–289. [Google Scholar] [CrossRef]
- Djuzenova, C.S.; Fischer, T.; Katzer, A.; Sisario, D.; Korsa, T.; Steussloff, G.; Sukhorukov, V.L.; Flentje, M. Opposite effects of the triple target (DNA-PK/PI3K/mTOR) inhibitor PI-103 on the radiation sensitivity of glioblastoma cell lines proficient and deficient in DNA-PKcs. BMC Cancer 2021, 21, 1201. [Google Scholar] [CrossRef]
- Haines, E.; Nishida, Y.; Carr, M.I.; Montoya, R.H.; Ostermann, L.B.; Zhang, W.; Zenke, F.T.; Blaukat, A.; Andreeff, M.; Vassilev, L.T. DNA-PK inhibitor peposertib enhances p53-dependent cytotoxicity of DNA double-strand break inducing therapy in acute leukemia. Sci. Rep. 2021, 11, 12148. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.I.; Zimmermann, A.; Chiu, L.Y.; Zenke, F.T.; Blaukat, A.; Vassilev, L.T. DNA-PK Inhibitor, M3814, as a New Combination Partner of Mylotarg in the Treatment of Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 127. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.C.; Enjeti, A.K.; Kahl, R.G.S.; Flanagan, H.M.; Sillar, J.; Skerrett-Byrne, D.A.; Al Mazi, J.G.; Au, G.G.; de Bock, C.E.; Evans, K.; et al. Quantitative phosphoproteomics uncovers synergy between DNA-PK and FLT3 inhibitors in acute myeloid leukaemia. Leukemia 2021, 35, 1782–1787. [Google Scholar] [CrossRef] [PubMed]
- Leahy, J.J.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Richardson, C.; Rigoreau, L.; Smith, G.C. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett. 2004, 14, 6083–6087. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Sapinoso, L.M.; Tran, T.; Gaffney, B.; Wong, L.; Sankar, S.; Raymon, H.K.; Mortensen, D.S.; Xu, S. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget 2017, 8, 74688–74702. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.J.; Laranjo, M.; Abrantes, A.M.; Casalta-Lopes, J.; Sarmento-Santos, D.; Costa, T.; Serambeque, B.; Almeida, N.; Goncalves, T.; Mamede, C.; et al. Endometrial Cancer Spheres Show Cancer Stem Cells Phenotype and Preference for Oxidative Metabolism. Pathol. Oncol. Res. 2019, 25, 1163–1174. [Google Scholar] [CrossRef]
- Costa, M.I.; Lapa, B.S.; Jorge, J.; Alves, R.; Carreira, I.M.; Sarmento-Ribeiro, A.B.; Goncalves, A.C. Zinc Prevents DNA Damage in Normal Cells but Shows Genotoxic and Cytotoxic Effects in Acute Myeloid Leukemia Cells. Int. J. Mol. Sci. 2022, 23, 2567. [Google Scholar] [CrossRef]
- Lapa, B.; Goncalves, A.C.; Jorge, J.; Alves, R.; Pires, A.S.; Abrantes, A.M.; Coucelo, M.; Abrunhosa, A.; Botelho, M.F.; Nascimento-Costa, J.M.; et al. Acute myeloid leukemia sensitivity to metabolic inhibitors: Glycolysis showed to be a better therapeutic target. Med. Oncol. 2020, 37, 72. [Google Scholar] [CrossRef]
- Nakamura, K.; Karmokar, A.; Farrington, P.M.; James, N.H.; Ramos-Montoya, A.; Bickerton, S.J.; Hughes, G.D.; Illidge, T.M.; Cadogan, E.B.; Davies, B.R.; et al. Inhibition of DNA-PK with AZD7648 Sensitizes Tumor Cells to Radiotherapy and Induces Type I IFN-Dependent Durable Tumor Control. Clin. Cancer Res. 2021, 27, 4353–4366. [Google Scholar] [CrossRef]
- Willoughby, C.E.; Jiang, Y.; Thomas, H.D.; Willmore, E.; Kyle, S.; Wittner, A.; Phillips, N.; Zhao, Y.; Tudhope, S.J.; Prendergast, L.; et al. Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J. Clin. Investig. 2020, 130, 258–271. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DDR Pathway | CML | AML | |||||
|---|---|---|---|---|---|---|---|
| K-562 | LAMA-84 | HEL | HL-60 | KG-1 | NB-4 | THP-1 | |
| Base excision repair | SMUG1 | MBD4 TDG | |||||
| Other base excision repair and strand break joining factors | APLF | PNKP | APLF | ||||
| Nucleotide excision repair | DDB1 GTF2H1 | ERCC5 XAB2 | RAD23A RPA3 | GTF2H4 | |||
| Nucleotide-excision-repair-related | ERCC6 | UVSSA | |||||
| Homologous recombination | EME1 | SPIDR | BARD1 RAD54B | SMC6 | BRAC1 PAXIP1 RAD51D RBBP8 | RAD51D | |
| Non-homologous end joining | PRKDC | PRKDC | PRKDC | XRCC5 | |||
| Fanconi anemia | FANCB FANCM | FANCD2 | BRAC2 FANCI | FANCD2 | FANCD2 FANCG PALB2 | ||
| Mismatch excision repair | MSH4 | MLH3 MSH6 | HFM1 | MLH3 MSH2 MSH3 PMS2 | |||
| Repair of DNA–protein crosslinks | TDP1 | ||||||
| DNA polymerases | POLI | POLA1 POLD1 | POLE POLN | REV3L | POLD2 POLQ REV1 | POLD3 POLM | |
| Chromatin structure and modification | CHAF1A | ||||||
| Editing and processing nucleases | DCLRE1A | ENDOV | DCLRE1A | ||||
| Modulation of nucleotide pools | NUDT1 | ||||||
| Other conserved DNA damage response genes | RAD17 TP53 | PER1 RAD1 TP53 | TP53 | delTP53* PER1 TP53BP1 | CHEK1 TOPBP1 TP53 | ATRIP CHEK1 MDC1 PER1 TP53 | TOPBP1 TP53 |
| Ubiquitination and modification | HERC2 | RNF8 | HERC2 | ||||
| Genes defective in diseases associated with sensitivity to DNA damaging agents | WRN | ATM TOP3A | |||||
| Other identified genes with known or suspected DNA repair function | PRPF19 | RECQL5 | |||||
| CML | AML | |||||||
|---|---|---|---|---|---|---|---|---|
| IMC | K-562 | LAMA-84 | HEL | HL-60 | KG-1 | NB-4 | THP-1 | |
| γH2AX expression (MIF) | 19.0 ± 1.5 | 37.0 ± 1.7 ** | 29.3 ± 3.3 | 66.3 ± 5.9 *** | 21.3 ± 2.2 | 25.3 ± 0.9 | 63.0 ± 1.5 *** | 30.3 ± 0.9 * |
| BN cells with CDBs (%) | 2.5 ± 0.2 | 7.2 ± 0.6 *** | 8.8 ± 0.8 *** | 3.6 ± 0.2 | 4.1 ± 0.3 | 5.4 ± 0.8 ** | 4.0 ± 0.3 | 5.9 ± 0.5 *** |
| Cells with MNi (%) | 57.8 ± 6.8 | 19.8 ± 2.9 *** | 24.5 ± 1.2 *** | 25.5 ± 4.2 *** | 30.9 ± 4.4 ** | 49.9 ± 5.0 | 30.4 ± 4.6 ** | 29.7 ± 7.1 ** |
| Cells with NBUDs (%) | 35.3 ± 6.3 | 52.2 ± 2.5 | 26.1 ± 2.3 | 52.8 ± 2.3 | 53.7 ± 4.0 | 27.5 ± 4.7 | 48.4 ± 4.0 | 57.9 ± 8.8 * |
| Cells with NPBs (%) | 6.9 ± 3.7 | 28.0 ± 2.6 ** | 49.4 ± 2.3 *** | 21.7 ± 4.3 * | 15.4 ± 4.1 | 22.6 ± 1.7 * | 21.2 ± 3.9 * | 12.4 ± 4.5 |
| Telomere length fold change (normalized to IMC cells) | 1 | 0.16 ± 0.01 *** | 0.25 ± 0.03 ** | 0.23 ± 0.04 ** | 0.16 ± 0.02 *** | 0.09 ± 0.01 *** | 0.26 ± 0.01 *** | 0.38 ± 0.02 *** |
| Sub G1 (%) | G0/G1 (%) | S (%) | G2/M (%) | ||
|---|---|---|---|---|---|
| K-562 | Control | 1.6 ± 0.9 | 36.8 ± 2.4 | 48.6 ± 3.1 | 14.6 ± 1.3 |
| AZD 200 µM | 4.2 ± 1.4 | 41.8 ± 3.5 | 49.4 ± 2.9 | 8.8 ± 1.7 * | |
| LAMA-84 | Control | 1.8 ± 1.0 | 57.6 ± 1.6 | 30.6 ± 1.9 | 11.8 ± 0.8 |
| AZD 100 µM | 4.6 ± 2.0 | 75.8 ± 1.5 *** | 17.6 ± 1.6 *** | 6.6 ± 0.8 | |
| AZD 200 µM | 5.4 ± 1.9 | 77.4 ± 1.3 *** | 18.2 ± 1.8 *** | 5.2 ± 0.8 ** | |
| HEL | Control | 1.4 ± 0.4 | 41.0 ± 2.6 | 38.2 ± 2.5 | 20.8 ± 0.9 |
| AZD 50 µM | 4.2 ± 0.9 | 71.4 ± 1.7 * | 24.8 ± 1.9 * | 3.8 ± 0.7 | |
| AZD 150 µM | 16.2 ± 2.8 ** | 72.0 ± 0.6 * | 27.2 ± 0.6 | 0.8 ± 0.4 ** | |
| HL-60 | Control | 0.6 ± 0.4 | 38.6 ± 2.4 | 46.2 ± 1.7 | 15.2 ± 1.5 |
| AZD 200 µM | 0.8 ± 0.2 | 83.2 ± 0.4 *** | 11.4 ± 0.4 ** | 5.4 ± 0.4 ** | |
| KG-1 | Control | 0.6 ± 0.2 | 44.8 ± 0.7 | 41.8 ± 0.7 | 13.4 ± 0.2 |
| AZD 100 µM | 1.2 ± 0.2 | 87.4 ± 0.4 *** | 7.2 ± 0.6 *** | 5.4 ± 0.2 *** | |
| AZD 200 µM | 2.2 ± 0.2 ** | 82.4 ± 0.9 | 10.0 ± 0.8 *** | 7.6 ± 0.2 | |
| NB-4 | Control | 0.2 ± 0.2 | 43.6 ± 1.6 | 48.4 ± 2.3 | 8.0 ± 1.9 |
| AZD 200 µM | 1.4 ± 0.5 | 83.4 ± 0.7 *** | 12.4 ± 0.6 *** | 4.2 ± 0.6 | |
| THP-1 | Control | 0.2 ± 0.2 | 49.8 ± 1.4 | 38.2 ± 1.3 | 12.0 ± 0.7 |
| AZD 200 µM | 1.6 ± 0.7 | 64.2 ± 0.7 *** | 27.4 ± 0.4 ** | 8.4 ± 0.5 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lapa, B.S.; Costa, M.I.; Figueiredo, D.; Jorge, J.; Alves, R.; Monteiro, A.R.; Serambeque, B.; Laranjo, M.; Botelho, M.F.; Carreira, I.M.; et al. AZD-7648, a DNA-PK Inhibitor, Induces DNA Damage, Apoptosis, and Cell Cycle Arrest in Chronic and Acute Myeloid Leukemia Cells. Int. J. Mol. Sci. 2023, 24, 15331. https://doi.org/10.3390/ijms242015331
Lapa BS, Costa MI, Figueiredo D, Jorge J, Alves R, Monteiro AR, Serambeque B, Laranjo M, Botelho MF, Carreira IM, et al. AZD-7648, a DNA-PK Inhibitor, Induces DNA Damage, Apoptosis, and Cell Cycle Arrest in Chronic and Acute Myeloid Leukemia Cells. International Journal of Molecular Sciences. 2023; 24(20):15331. https://doi.org/10.3390/ijms242015331
Chicago/Turabian StyleLapa, Beatriz Santos, Maria Inês Costa, Diana Figueiredo, Joana Jorge, Raquel Alves, Ana Raquel Monteiro, Beatriz Serambeque, Mafalda Laranjo, Maria Filomena Botelho, Isabel Marques Carreira, and et al. 2023. "AZD-7648, a DNA-PK Inhibitor, Induces DNA Damage, Apoptosis, and Cell Cycle Arrest in Chronic and Acute Myeloid Leukemia Cells" International Journal of Molecular Sciences 24, no. 20: 15331. https://doi.org/10.3390/ijms242015331
APA StyleLapa, B. S., Costa, M. I., Figueiredo, D., Jorge, J., Alves, R., Monteiro, A. R., Serambeque, B., Laranjo, M., Botelho, M. F., Carreira, I. M., Sarmento-Ribeiro, A. B., & Gonçalves, A. C. (2023). AZD-7648, a DNA-PK Inhibitor, Induces DNA Damage, Apoptosis, and Cell Cycle Arrest in Chronic and Acute Myeloid Leukemia Cells. International Journal of Molecular Sciences, 24(20), 15331. https://doi.org/10.3390/ijms242015331