
Sleeping Beauty Transposon Insertions into Nucleolar DNA by an Engineered Transposase Localized in the Nucleolus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
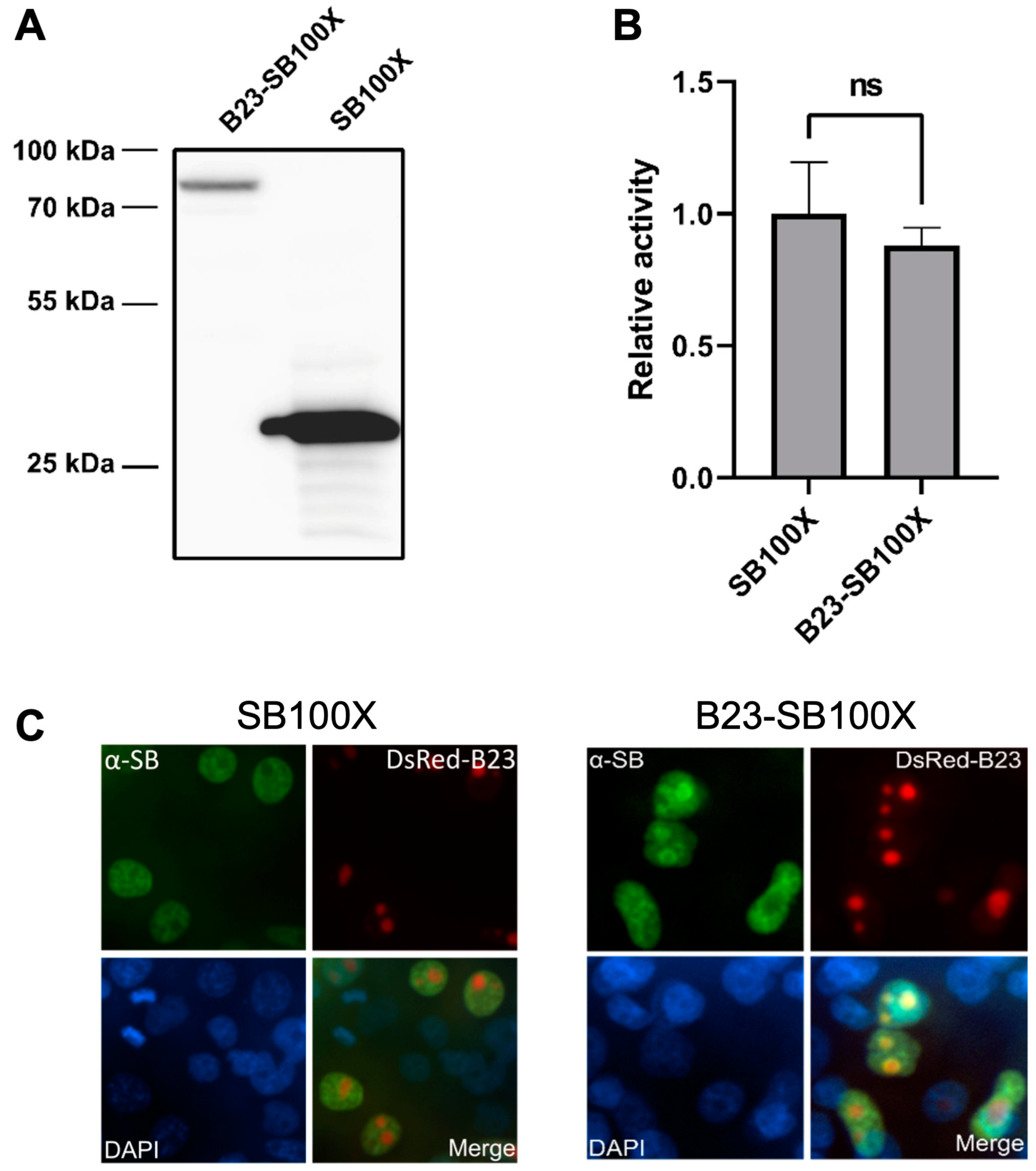
2.1. A B23-SB100X Fusion Protein Retains Transpositional Activity and Is Localized to the Nucleolus
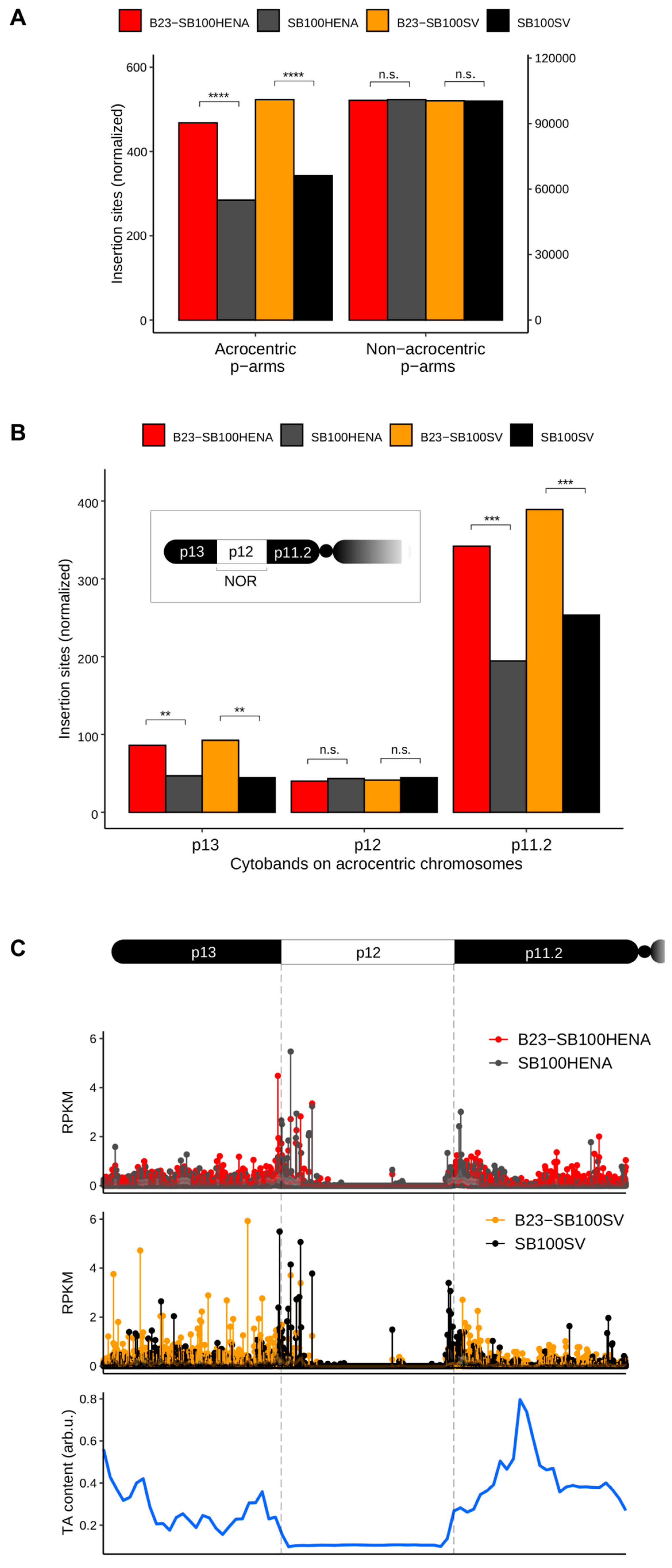
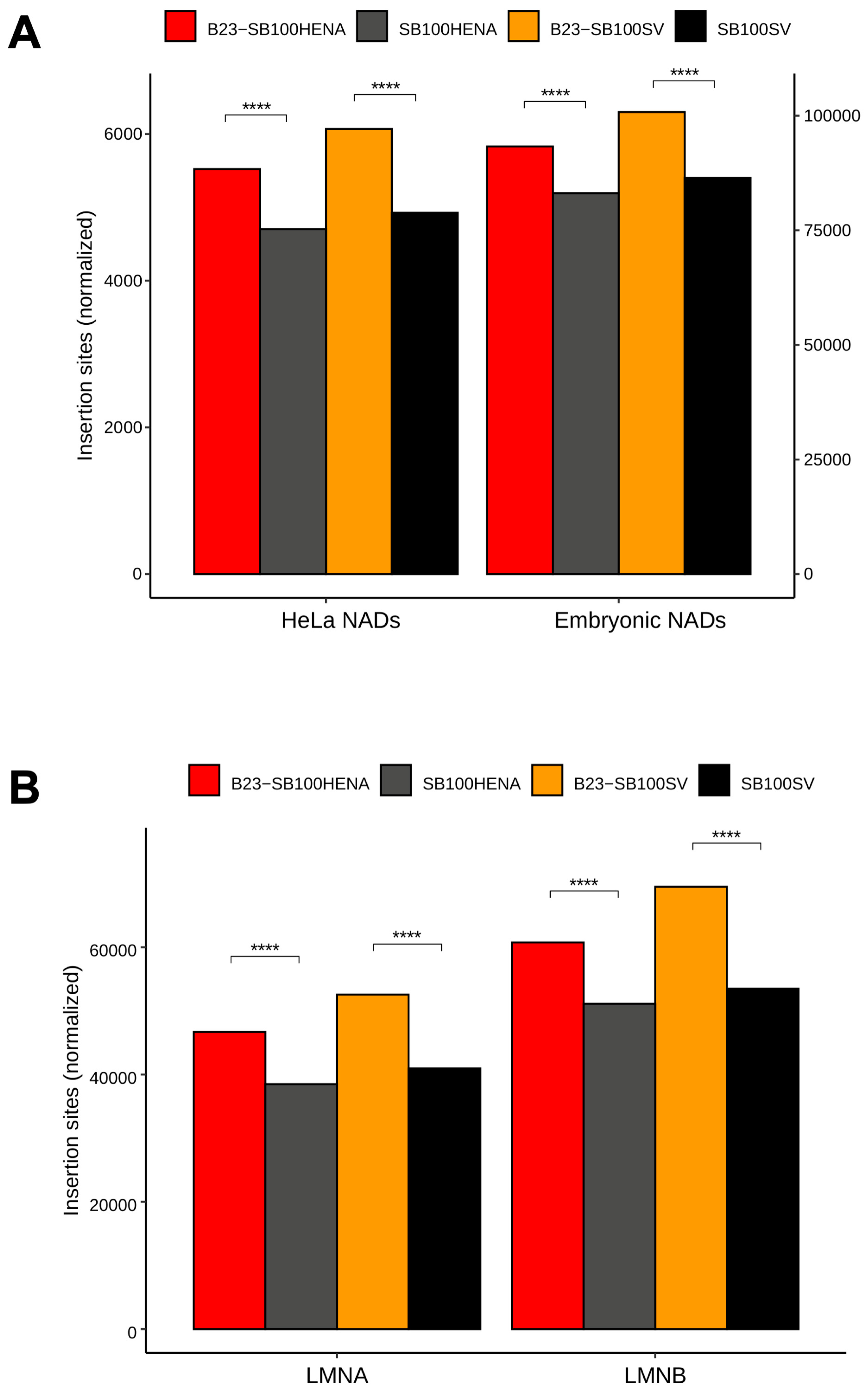
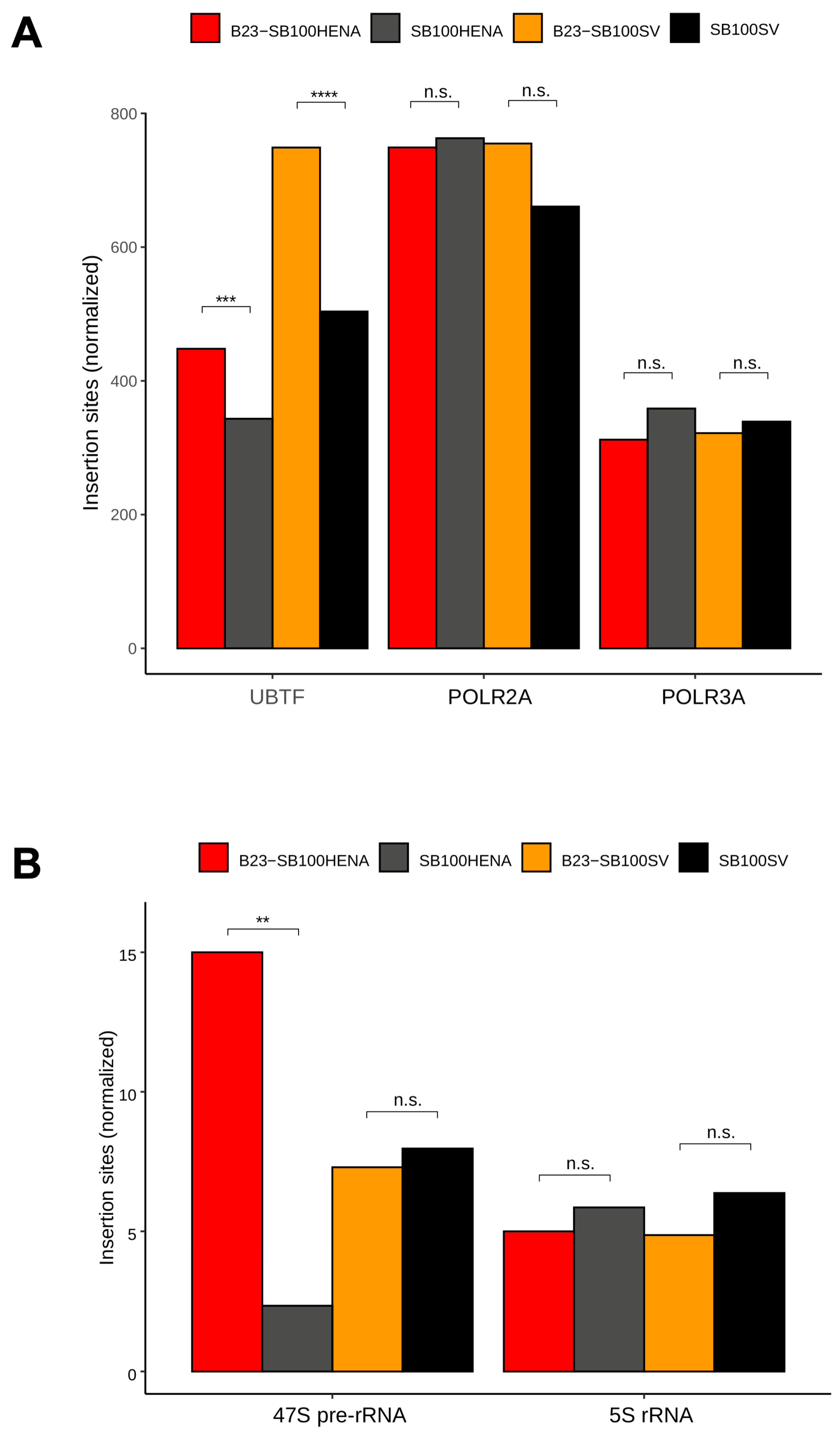
2.2. Sleeping Beauty Transposon Integrations Catalyzed by B23-SB100X Preferentially Occur within and in the Vicinity of Nucleolar Chromatin
3. Discussion
4. Materials and Methods
4.1. Generation of the B23-SB100X Fusion
4.2. Western Blot
4.3. Colony Formation Assay
4.4. Immunofluorescence Microscopy
4.5. Generation of Integration Libraries
4.6. Integration Site Sequencing and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Carusillo, A.; Mussolino, C. DNA Damage: From Threat to Treatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, G.; Andersen, T.; Zhou, P.; Pu, W.T. Optimization of genome engineering approaches with the CRISPR/Cas9 system. PLoS ONE 2014, 9, e105779. [Google Scholar] [CrossRef]
- Fung, H.; Weinstock, D.M. Repair at single targeted DNA double-strand breaks in pluripotent and differentiated human cells. PLoS ONE 2011, 6, e20514. [Google Scholar] [CrossRef]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi-Iwai, Y.; Shinohara, A.; Takeda, S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998, 17, 5497–5508. [Google Scholar] [CrossRef]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Ochmann, M.T.; Ivics, Z. Jumping Ahead with Sleeping Beauty: Mechanistic Insights into Cut-and-Paste Transposition. Viruses 2021, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Sultana, T.; Zamborlini, A.; Cristofari, G.; Lesage, P. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017, 18, 292–308. [Google Scholar] [CrossRef]
- Kovač, A.; Ivics, Z. Specifically integrating vectors for targeted gene delivery: Progress and prospects. Cell Gene Ther. Insights 2017, 3, 103–123. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Cavazza, A.; Moiani, A.; Mavilio, F. Mechanisms of retroviral integration and mutagenesis. Hum. Gene Ther. 2013, 24, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvak, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Ivics, Z.; Li, M.A.; Mates, L.; Boeke, J.D.; Nagy, A.; Bradley, A.; Izsvak, Z. Transposon-mediated genome manipulation in vertebrates. Nat. Methods 2009, 6, 415–422. [Google Scholar] [CrossRef]
- Hackett, P.B.; Largaespada, D.A.; Cooper, L.J.N. A transposon and transposase system for human application. Mol. Ther. 2010, 18, 674–683. [Google Scholar] [CrossRef]
- Boehme, P.; Doerner, J.; Solanki, M.; Jing, L.; Zhang, W.; Ehrhardt, A. The sleeping beauty transposon vector system for treatment of rare genetic diseases: An unrealized hope? Curr. Gene Ther. 2015, 15, 255–265. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Adams, D.J.; Wong, C.C. The utility of transposon mutagenesis for cancer studies in the era of genome editing. Genome Biol. 2015, 16, 229. [Google Scholar] [CrossRef]
- Hudecek, M.; Ivics, Z. Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr. Opin. Genet. Dev. 2018, 52, 100–108. [Google Scholar] [CrossRef]
- Hudecek, M.; Izsvak, Z.; Johnen, S.; Renner, M.; Thumann, G.; Ivics, Z. Going non-viral: The Sleeping Beauty transposon system breaks on through to the clinical side. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 1–26. [Google Scholar] [CrossRef]
- Kawakami, K.; Largaespada, D.A.; Ivics, Z. Transposons As Tools for Functional Genomics in Vertebrate Models. Trends Genet. 2017, 33, 784–801. [Google Scholar] [CrossRef] [PubMed]
- Amberger, M.; Ivics, Z. Latest Advances for the Sleeping Beauty Transposon System: 23 Years of Insomnia but Prettier than Ever: Refinement and Recent Innovations of the Sleeping Beauty Transposon System Enabling Novel, Nonviral Genetic Engineering Applications. Bioessays 2020, 42, e2000136. [Google Scholar] [CrossRef]
- Kebriaei, P.; Izsvák, Z.; Narayanavari, S.A.; Singh, H.; Ivics, Z. Gene Therapy with the Sleeping Beauty Transposon System. Trends Genet. 2017, 33, 852–870. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, P.J.; Largaespada, D.A. Transposon Insertion Mutagenesis in Mice for Modeling Human Cancers: Critical Insights Gained and New Opportunities. Int. J. Mol. Sci. 2020, 21, 1172. [Google Scholar] [CrossRef] [PubMed]
- de Jong, J.; Akhtar, W.; Badhai, J.; Rust, A.G.; Rad, R.; Hilkens, J.; Berns, A.; van Lohuizen, M.; Wessels, L.F.; de Ridder, J. Chromatin landscapes of retroviral and transposon integration profiles. PLoS Genet. 2014, 10, e1004250. [Google Scholar] [CrossRef] [PubMed]
- Yant, S.R.; Wu, X.; Huang, Y.; Garrison, B.; Burgess, S.M.; Kay, M.A. High-resolution genome-wide mapping of transposon integration in mammals. Mol. Cell Biol. 2005, 25, 2085–2094. [Google Scholar] [CrossRef]
- Moldt, B.; Miskey, C.; Staunstrup, N.H.; Gogol-Doring, A.; Bak, R.O.; Sharma, N.; Mates, L.; Izsvak, Z.; Chen, W.; Ivics, Z.; et al. Comparative genomic integration profiling of Sleeping Beauty transposons mobilized with high efficacy from integrase-defective lentiviral vectors in primary human cells. Mol. Ther. 2011, 19, 1499–1510. [Google Scholar] [CrossRef]
- Li, X.; Ewis, H.; Hice, R.H.; Malani, N.; Parker, N.; Zhou, L.; Feschotte, C.; Bushman, F.D.; Atkinson, P.W.; Craig, N.L. A resurrected mammalian hAT transposable element and a closely related insect element are highly active in human cell culture. Proc. Natl. Acad. Sci. USA 2013, 110, E478–E487. [Google Scholar] [CrossRef]
- Gogol-Doring, A.; Ammar, I.; Gupta, S.; Bunse, M.; Miskey, C.; Chen, W.; Uckert, W.; Schulz, T.F.; Izsvak, Z.; Ivics, Z. Genome-wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the piggyBac Transposon in Primary Human CD4(+) T Cells. Mol. Ther. 2016, 24, 592–606. [Google Scholar] [CrossRef]
- El Ashkar, S.; De Rijck, J.; Demeulemeester, J.; Vets, S.; Madlala, P.; Cermakova, K.; Debyser, Z.; Gijsbers, R. BET-independent MLV-based Vectors Target Away From Promoters and Regulatory Elements. Mol. Ther. Nucleic Acids 2014, 3, e179. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Ousterout, D.G.; Gersbach, C.A. The Development of TALE Nucleases for Biotechnology. Methods Mol. Biol. 2016, 1338, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Katzer, A.; Stuwe, E.E.; Fiedler, D.; Knespel, S.; Izsvak, Z. Targeted Sleeping Beauty transposition in human cells. Mol. Ther. 2007, 15, 1137–1144. [Google Scholar] [CrossRef]
- Ammar, I.; Gogol-Doring, A.; Miskey, C.; Chen, W.; Cathomen, T.; Izsvak, Z.; Ivics, Z. Retargeting transposon insertions by the adeno-associated virus Rep protein. Nucleic Acids Res. 2012, 40, 6693–6712. [Google Scholar] [CrossRef] [PubMed]
- Voigt, K.; Gogol-Doring, A.; Miskey, C.; Chen, W.; Cathomen, T.; Izsvak, Z.; Ivics, Z. Retargeting sleeping beauty transposon insertions by engineered zinc finger DNA-binding domains. Mol. Ther. 2012, 20, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Galvan, D.L.; Woodard, L.E.; Dorset, D.; Levy, S.; Wilson, M.H. Comparative analysis of chimeric ZFP-, TALE- and Cas9-piggyBac transposases for integration into a single locus in human cells. Nucleic Acids Res. 2017, 45, 8411–8422. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Chalmers, R. Targeted DNA transposition in vitro using a dCas9-transposase fusion protein. Nucleic Acids Res. 2019, 47, 8126–8135. [Google Scholar] [CrossRef] [PubMed]
- Hew, B.E.; Sato, R.; Mauro, D.; Stoytchev, I.; Owens, J.B. RNA-guided piggyBac transposition in human cells. Synth. Biol. 2019, 4, ysz018. [Google Scholar] [CrossRef]
- Kovač, A.; Miskey, C.; Menzel, M.; Grueso, E.; Gogol-Döring, A.; Ivics, Z. RNA-guided retargeting of Sleeping Beauty transposition in human cells. eLife 2020, 9, e53868. [Google Scholar] [CrossRef]
- Goshayeshi, L.; Yousefi Taemeh, S.; Dehdilani, N.; Nasiri, M.; Ghahramani Seno, M.M.; Dehghani, H. CRISPR/dCas9-mediated transposition with specificity and efficiency of site-directed genomic insertions. FASEB J. 2021, 35, e21359. [Google Scholar] [CrossRef]
- Zhang, X.; Eickbush, M.T.; Eickbush, T.H. Role of recombination in the long-term retention of transposable elements in rRNA gene loci. Genetics 2008, 180, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Eickbush, M.T.; Eickbush, T.H. A population genetic model for the maintenance of R2 retrotransposons in rRNA gene loci. PLoS Genet. 2013, 9, e1003179. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Penton, E.H.; Sullender, B.W.; Crease, T.J. Pokey, a new DNA transposon in Daphnia (cladocera: Crustacea). J. Mol. Evol. 2002, 55, 664–673. [Google Scholar] [CrossRef]
- Elliott, T.A.; Stage, D.E.; Crease, T.J.; Eickbush, T.H. In and out of the rRNA genes: Characterization of Pokey elements in the sequenced Daphnia genome. Mob. DNA 2013, 4, 20. [Google Scholar] [CrossRef]
- Eagle, S.H.C.; Crease, T.J. Distribution of the DNA transposon family, Pokey in the Daphnia pulex species complex. Mob. DNA 2016, 7, 11. [Google Scholar] [CrossRef]
- Eickbush, T. Fruit flies and humans respond differently to retrotransposons. Curr. Opin. Genet. Dev. 2002, 12, 669–674. [Google Scholar] [CrossRef]
- McClintock, B. The relationship of a particular chromosomal element to the development of the nucleoli in Zea Mays. Z. Zellforsch. Mikrosk. Anat. 1934, 21, 294–328. [Google Scholar] [CrossRef]
- McStay, B. Nucleolar organizer regions: Genomic ’dark matter’ requiring illumination. Genes. Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef]
- Floutsakou, I.; Agrawal, S.; Nguyen, T.T.; Seoighe, C.; Ganley, A.R.D.; McStay, B. The shared genomic architecture of human nucleolar organizer regions. Genome Res. 2013, 23, 2003–2012. [Google Scholar] [CrossRef]
- Andersen, J.S.; Lam, Y.W.; Leung, A.K.L.; Ong, S.-E.; Lyon, C.E.; Lamond, A.I.; Mann, M. Nucleolar proteome dynamics. Nature 2005, 433, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Ciganda, M.; Williams, N. Eukaryotic 5S rRNA biogenesis. Wiley Interdiscip. Rev. RNA 2011, 2, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Schöfer, C.; Weipoltshammer, K. Nucleolus and chromatin. Histochem. Cell Biol. 2018, 150, 209–225. [Google Scholar] [CrossRef]
- Boisvert, F.M.; van Koningsbruggen, S.; Navascues, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef]
- van de Nobelen, S.; Rosa-Garrido, M.; Leers, J.; Heath, H.; Soochit, W.; Joosen, L.; Jonkers, I.; Demmers, J.; van der Reijden, M.; Torrano, V.; et al. CTCF regulates the local epigenetic state of ribosomal DNA repeats. Epigenetics Chromatin 2010, 3, 19. [Google Scholar] [CrossRef]
- Wang, Z.; Lisowski, L.; Finegold, M.J.; Nakai, H.; Kay, M.A.; Grompe, M. AAV vectors containing rDNA homology display increased chromosomal integration and transgene persistence. Mol. Ther. 2012, 20, 1902–1911. [Google Scholar] [CrossRef]
- Lisowski, L.; Lau, A.; Wang, Z.; Zhang, Y.; Zhang, F.; Grompe, M.; Kay, M.A. Ribosomal DNA integrating rAAV-rDNA vectors allow for stable transgene expression. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1912–1923. [Google Scholar] [CrossRef]
- Johnson, J.S.; Samulski, R.J. Enhancement of adeno-associated virus infection by mobilizing capsids into and out of the nucleolus. J. Virol. 2009, 83, 2632–2644. [Google Scholar] [CrossRef]
- Nakai, H.; Wu, X.; Fuess, S.; Storm, T.A.; Munroe, D.; Montini, E.; Burgess, S.M.; Grompe, M.; Kay, M.A. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J. Virol. 2005, 79, 3606–3614. [Google Scholar] [CrossRef]
- Miller, D.G.; Trobridge, G.D.; Petek, L.M.; Jacobs, M.A.; Kaul, R.; Russell, D.W. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J. Virol. 2005, 79, 11434–11442. [Google Scholar] [CrossRef] [PubMed]
- Schenkwein, D.; Turkki, V.; Ahlroth, M.K.; Timonen, O.; Airenne, K.J.; Yla-Herttuala, S. rDNA-directed integration by an HIV-1 integrase—I-PpoI fusion protein. Nucleic Acids Res. 2013, 41, e61. [Google Scholar] [CrossRef]
- Bire, S.; Dusserre, Y.; Bigot, Y.; Mermod, N. PiggyBac transposase and transposon derivatives for gene transfer targeting the ribosomal DNA loci of CHO cells. J. Biotechnol. 2021, 341, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Shida, H.; Maki, M.; Hatanaka, M. Effects of a highly basic region of human immunodeficiency virus Tat protein on nucleolar localization. J. Virol. 1990, 64, 1803–1807. [Google Scholar] [CrossRef]
- Cochrane, A.W.; Perkins, A.; Rosen, C.A. Identification of sequences important in the nucleolar localization of human immunodeficiency virus Rev: Relevance of nucleolar localization to function. J. Virol. 1990, 64, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Valdez, B.C.; Perlaky, L.; Henning, D.; Saijo, Y.; Chan, P.K.; Busch, H. Identification of the nuclear and nucleolar localization signals of the protein p120. Interaction with translocation protein B23. J. Biol. Chem. 1994, 269, 23776–23783. [Google Scholar] [CrossRef]
- Siomi, H.; Shida, H.; Nam, S.H.; Nosaka, T.; Maki, M.; Hatanaka, M. Sequence requirements for nucleolar localization of human T cell leukemia virus type I pX protein, which regulates viral RNA processing. Cell 1988, 55, 197–209. [Google Scholar] [CrossRef]
- Lopez, D.J.; Rodriguez, J.A.; Banuelos, S. Nucleophosmin, a multifunctional nucleolar organizer with a role in DNA repair. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140532. [Google Scholar] [CrossRef]
- Louvet, E.; Junera, H.R.; Berthuy, I.; Hernandez-Verdun, D. Compartmentation of the nucleolar processing proteins in the granular component is a CK2-driven process. Mol. Biol. Cell 2006, 17, 2537–2546. [Google Scholar] [CrossRef]
- Yant, S.R.; Huang, Y.; Akache, B.; Kay, M.A. Site-directed transposon integration in human cells. Nucleic Acids Res. 2007, 35, e50. [Google Scholar] [CrossRef]
- Ivics, Z.; Izsvak, Z.; Minter, A.; Hackett, P.B. Identification of functional domains and evolution of Tc1-like transposable elements. Proc. Natl. Acad. Sci. USA 1996, 93, 5008–5013. [Google Scholar] [CrossRef]
- Palmer, T.D.; Miller, A.D.; Reeder, R.H.; McStay, B. Efficient expression of a protein coding gene under the control of an RNA polymerase I promoter. Nucleic Acids Res. 1993, 21, 3451–3457. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Vigdal, T.J.; Kaufman, C.D.; Izsvak, Z.; Voytas, D.F.; Ivics, Z. Common physical properties of DNA affecting target site selection of sleeping beauty and other Tc1/mariner transposable elements. J. Mol. Biol. 2002, 323, 441–452. [Google Scholar] [CrossRef]
- Nemeth, A.; Langst, G. Genome organization in and around the nucleolus. Trends Genet. 2011, 27, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Briand, N.; Collas, P. Lamina-associated domains: Peripheral matters and internal affairs. Genome Biol. 2020, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2012, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Lee, G.; Malani, N.; Setty, M.; Riviere, I.; Tirunagari, L.M.; Kadota, K.; Roth, S.L.; Giardina, P.; Viale, A.; et al. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 73–78. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Schambach, A. Gene Insertion Into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 2016, 24, 678–684. [Google Scholar] [CrossRef]
- Xiong, Y.E.; Eickbush, T.H. Functional expression of a sequence-specific endonuclease encoded by the retrotransposon R2Bm. Cell 1988, 55, 235–246. [Google Scholar] [CrossRef]
- Feng, Q.; Schumann, G.; Boeke, J.D. Retrotransposon R1Bm endonuclease cleaves the target sequence. Proc. Natl. Acad. Sci. USA 1998, 95, 2083–2088. [Google Scholar] [CrossRef] [PubMed]
- Heliot, L.; Mongelard, F.; Klein, C.; O’Donohue, M.F.; Chassery, J.M.; Robert-Nicoud, M.; Usson, Y. Nonrandom distribution of metaphase AgNOR staining patterns on human acrocentric chromosomes. J. Histochem. Cytochem. 2000, 48, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Roussel, P.; Andre, C.; Comai, L.; Hernandez-Verdun, D. The rDNA transcription machinery is assembled during mitosis in active NORs and absent in inactive NORs. J. Cell Biol. 1996, 133, 235–246. [Google Scholar] [CrossRef]
- Preuss, S.; Pikaard, C.S. rRNA gene silencing and nucleolar dominance: Insights into a chromosome-scale epigenetic on/off switch. Biochim. Biophys. Acta 2007, 1769, 383–392. [Google Scholar] [CrossRef]
- Warsinger-Pepe, N.; Li, D.; Yamashita, Y.M. Regulation of Nucleolar Dominance in Drosophila melanogaster. Genetics 2020, 214, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Grabundzija, I.; Irgang, M.; Mates, L.; Belay, E.; Matrai, J.; Gogol-Doring, A.; Kawakami, K.; Chen, W.; Ruiz, P.; Chuah, M.K.; et al. Comparative analysis of transposable element vector systems in human cells. Mol. Ther. 2010, 18, 1200–1209. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Teissandier, A.; Servant, N.; Barillot, E.; Bourc’his, D. Tools and best practices for retrotransposon analysis using high-throughput sequencing data. Mob. DNA 2019, 10, 52. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Akalin, A.; Franke, V.; Vlahovicek, K.; Mason, C.E.; Schubeler, D. Genomation: A toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 2015, 31, 1127–1129. [Google Scholar] [CrossRef]
- Dillinger, S.; Straub, T.; Nemeth, A. Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS ONE 2017, 12, e0178821. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Conesa, A.; Santoyo-Lopez, J.; Medina, I.; Montaner, D.; Peterfia, B.; Solovei, I.; Cremer, T.; Dopazo, J.; Langst, G. Initial genomics of the human nucleolus. PLoS Genet. 2010, 6, e1000889. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.G.; Duband-Goulet, I.; Oldenburg, A.; Buendia, B.; Collas, P. Distinct features of lamin A-interacting chromatin domains mapped by ChIP-sequencing from sonicated or micrococcal nuclease-digested chromatin. Nucleus 2015, 6, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovač, A.; Miskey, C.; Ivics, Z. Sleeping Beauty Transposon Insertions into Nucleolar DNA by an Engineered Transposase Localized in the Nucleolus. Int. J. Mol. Sci. 2023, 24, 14978. https://doi.org/10.3390/ijms241914978
Kovač A, Miskey C, Ivics Z. Sleeping Beauty Transposon Insertions into Nucleolar DNA by an Engineered Transposase Localized in the Nucleolus. International Journal of Molecular Sciences. 2023; 24(19):14978. https://doi.org/10.3390/ijms241914978
Chicago/Turabian StyleKovač, Adrian, Csaba Miskey, and Zoltán Ivics. 2023. "Sleeping Beauty Transposon Insertions into Nucleolar DNA by an Engineered Transposase Localized in the Nucleolus" International Journal of Molecular Sciences 24, no. 19: 14978. https://doi.org/10.3390/ijms241914978