
Robust AMBER Force Field Parameters for Glutathionylated Cysteines

Abstract
:1. Introduction
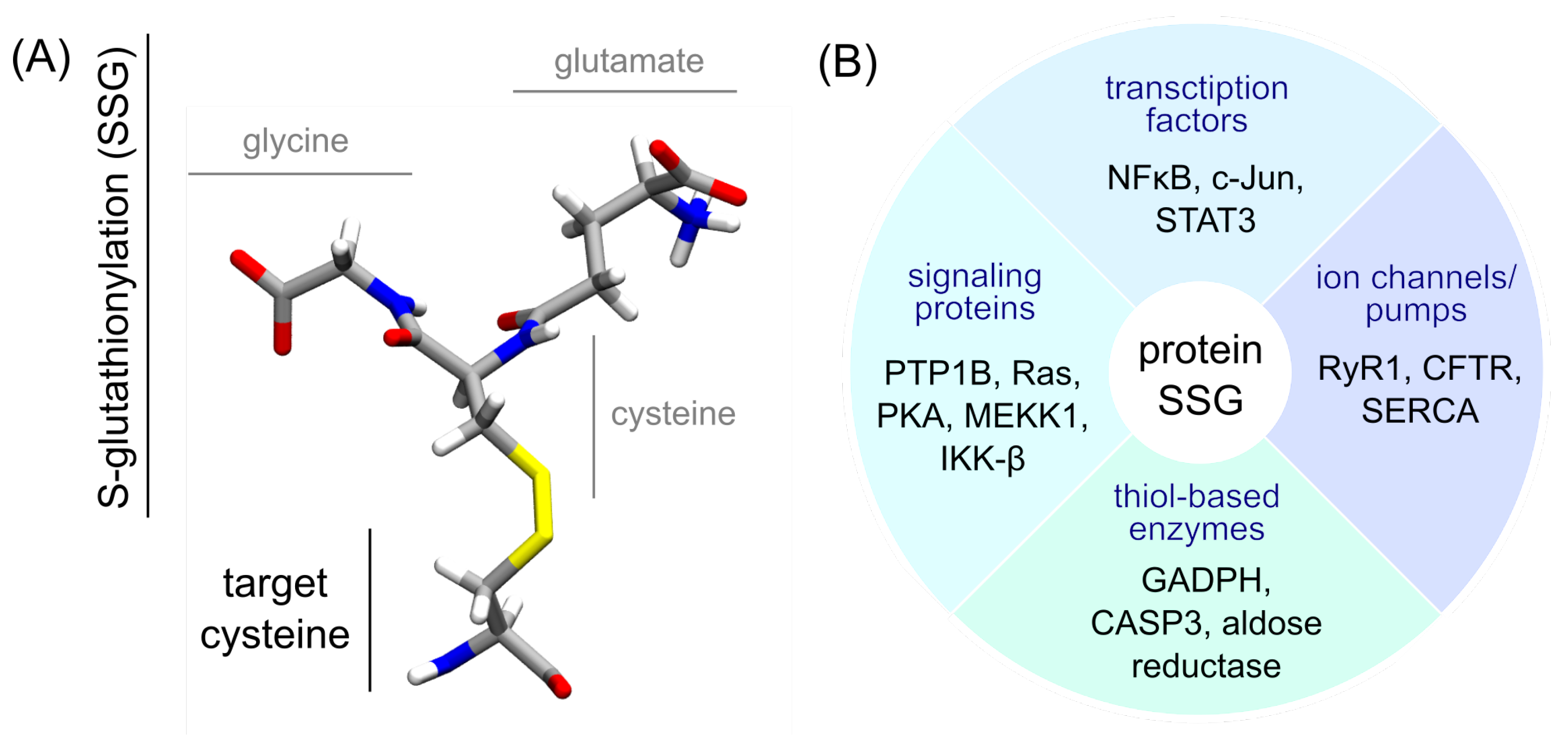
2. Results
2.1. Experimental Structure Curation
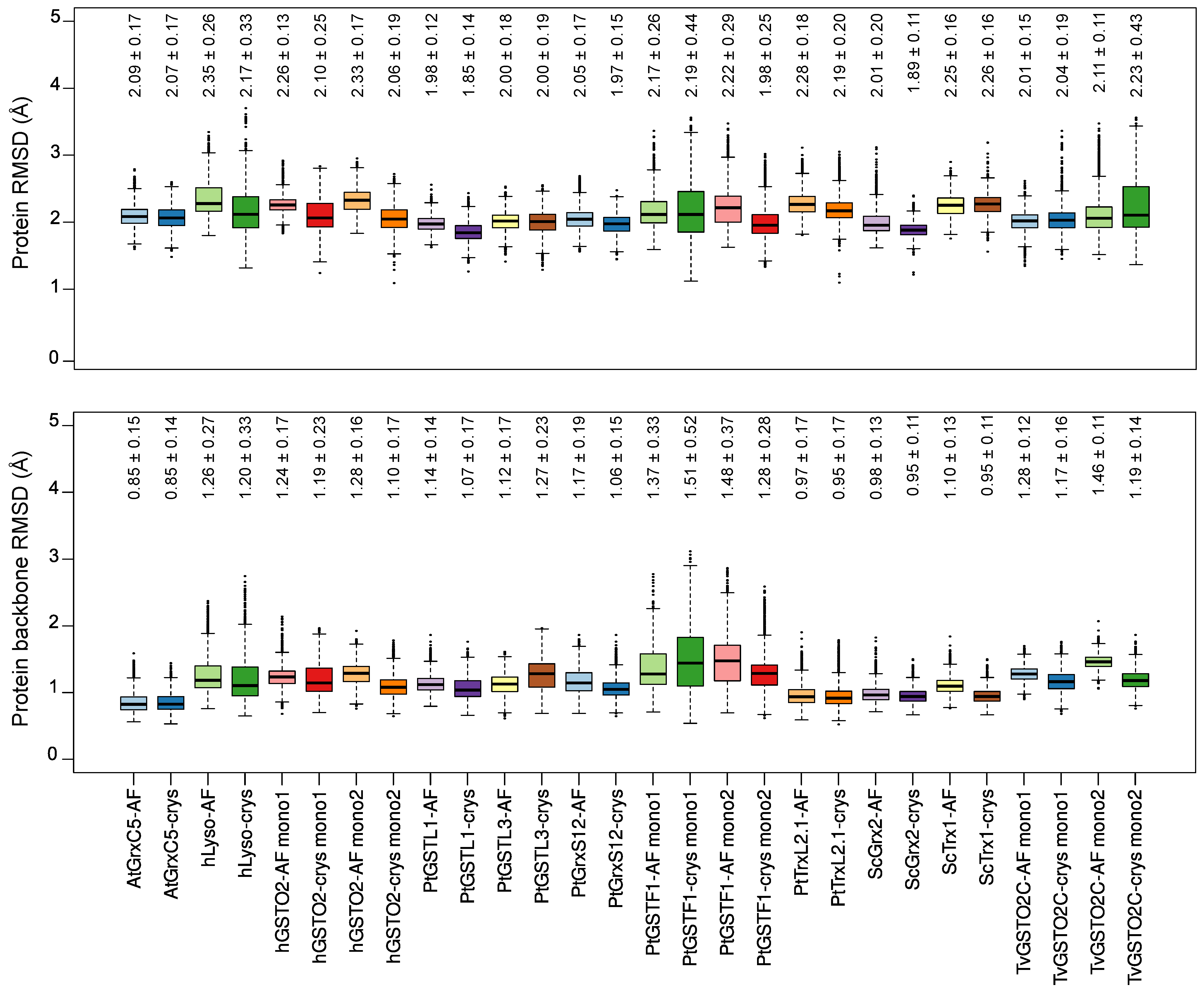
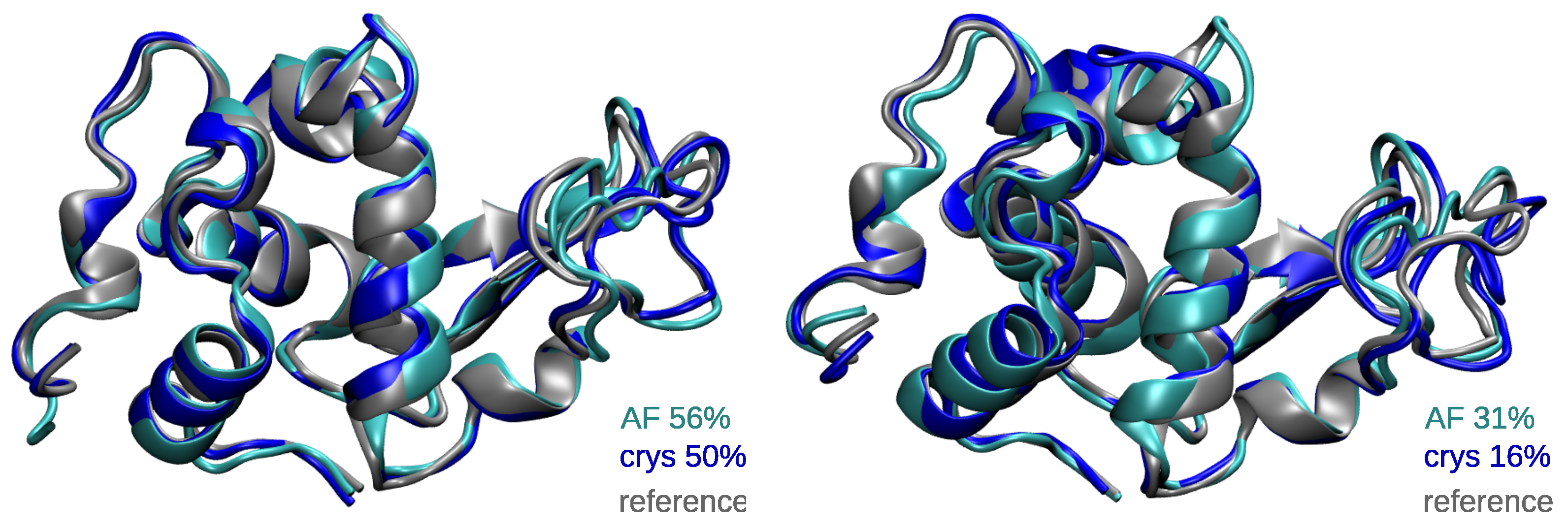
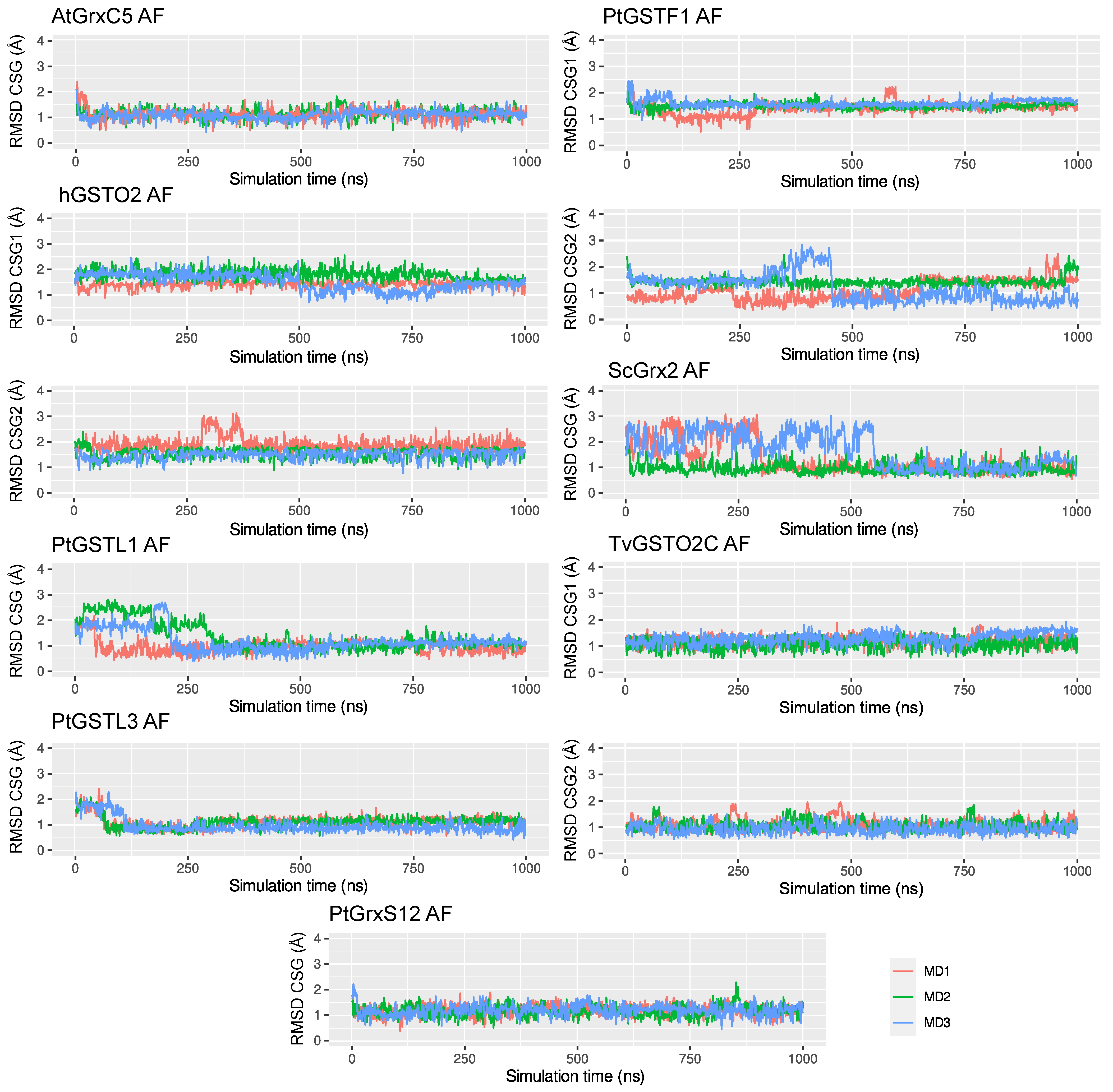
2.2. Global Structural Features of the Systems
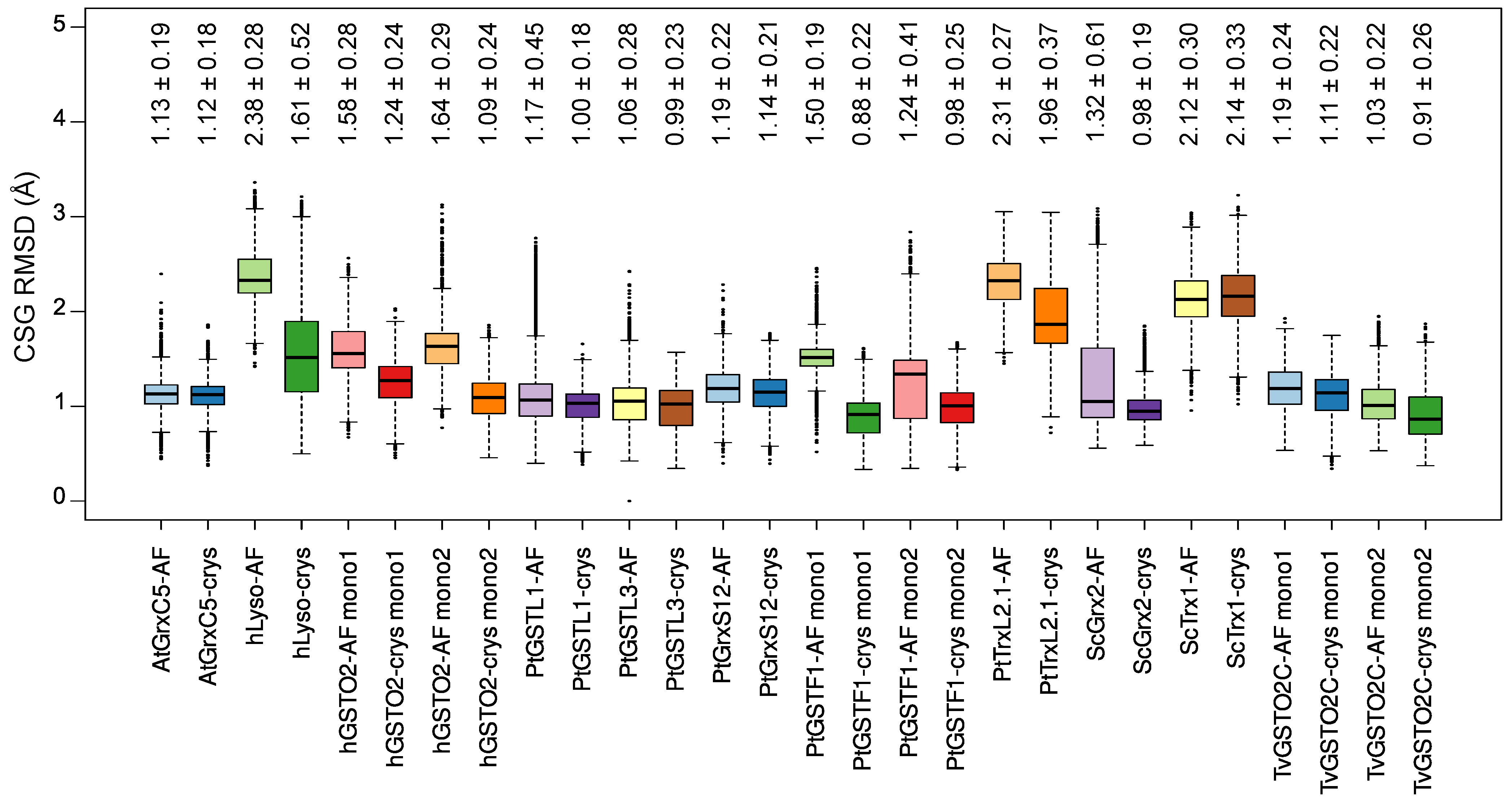
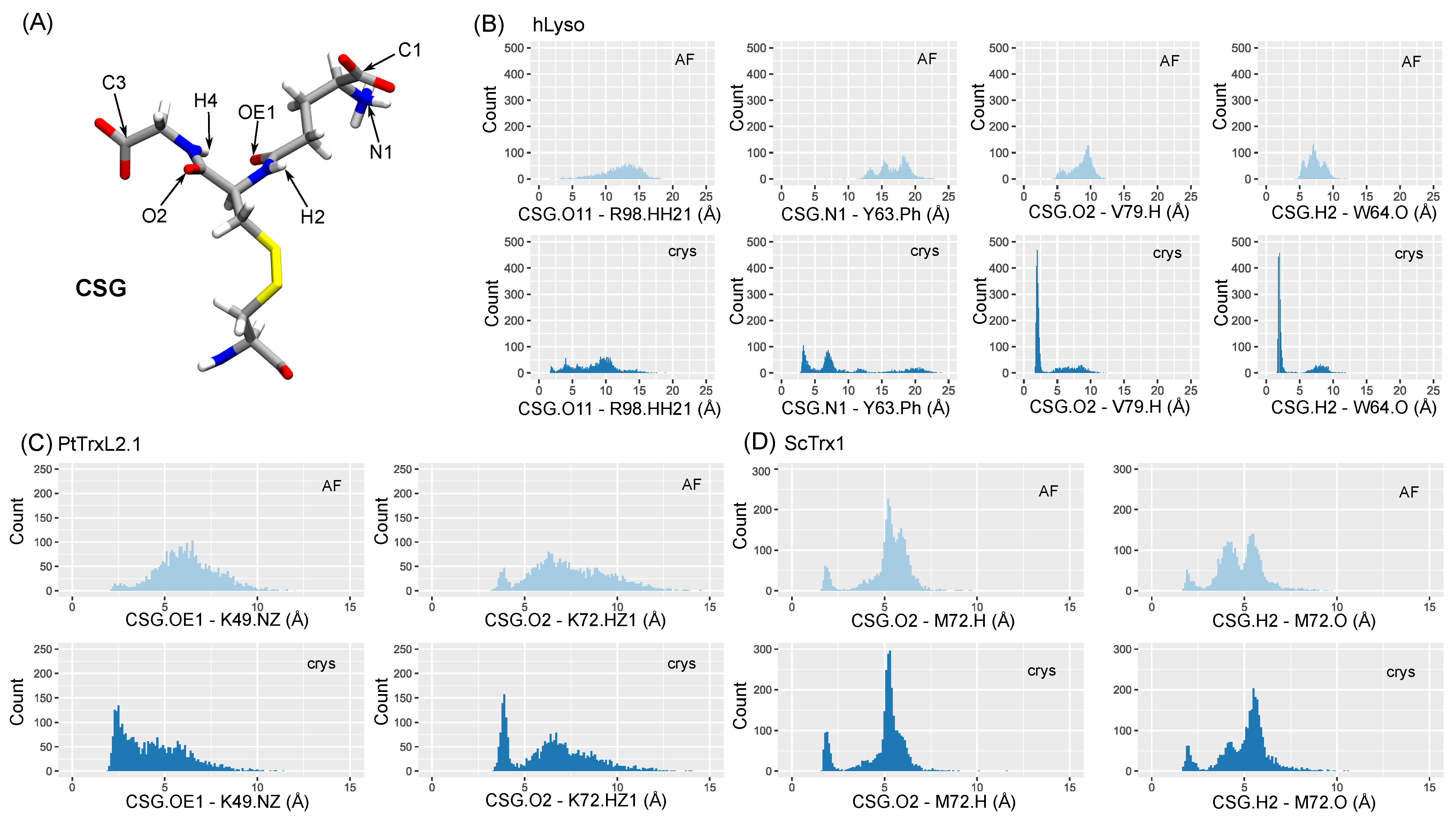
2.3. CSG Structural Features
2.4. CSG Interaction Network
3. Discussion
4. Materials and Methods
4.1. Generation of the CSG Parameters
4.2. System Setup
4.3. Molecular Dynamics Simulations Protocol
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Cell signalling and the glutathione redox system. Biochem. Pharmacol. 2002, 64, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Anathy, V.; Roberson, E.C.; Guala, A.S.; Godburn, K.E.; Budd, R.C.; Janssen-Heininger, Y.M. Redox-based regulation of apoptosis: S-glutathionylation as a regulatory mechanism to control cell death. Antioxid. Redox Signal. 2012, 16, 496–505. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Olaso, G.; Hake, S.B.; Bönisch, C.; Wiedemann, S.M.; Markovic, J.; Dasi, F.; Gimeno, A.; Pérez-Quilis, C.; Palacios, O.; et al. Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid. Redox Signal. 2013, 19, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.V.; Novichkova, M.D. S-Glutathionylation and S-Nitrosylation as Modulators of Redox-Dependent Processes in Cancer Cell. Biochemistry 2023, 88, 924–943. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.w.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef]
- Manevich, Y.; Feinstein, S.; Fisher, A. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with πGST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.H.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernández, J.M. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Checconi, P.; Limongi, D.; Baldelli, S.; Ciriolo, M.R.; Nencioni, L.; Palamara, A.T. Role of glutathionylation in infection and inflammation. Nutrients 2019, 11, 1952. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M. S-glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Mol. Interv. 2007, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed]
- Musaogullari, A.; Chai, Y.C. Redox regulation by protein S-glutathionylation: From molecular mechanisms to implications in health and disease. Int. J. Mol. Sci. 2020, 21, 8113. [Google Scholar] [CrossRef]
- Pal, D.; Rai, A.; Checker, R.; Patwardhan, R.; Singh, B.; Sharma, D.; Sandur, S.K. Role of protein S-Glutathionylation in cancer progression and development of resistance to anti-cancer drugs. Arch. Biochem. Biophys. 2021, 704, 108890. [Google Scholar] [CrossRef]
- Oppong, D.; Schiff, W.; Shivamadhu, M.C.; Ahn, Y.H. Chemistry and biology of enzymes in protein glutathionylation. Curr. Opin. Chem. Biol. 2023, 75, 102326. [Google Scholar] [CrossRef]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef]
- Xie, Y.; Kole, S.; Precht, P.; Pazin, M.J.; Bernier, M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 2009, 150, 1122–1131. [Google Scholar] [CrossRef]
- Weigle, A.T.; Feng, J.; Shukla, D. Thirty years of molecular dynamics simulations on posttranslational modifications of proteins. Phys. Chem. Chem. Phys. 2022, 24, 26371–26397. [Google Scholar] [CrossRef]
- Hyslop, P.A.; Boggs, L.N.; Chaney, M.O. Origin of Elevated S-Glutathionylated GAPDH in Chronic Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 5529. [Google Scholar] [CrossRef]
- Moffett, A.S.; Bender, K.W.; Huber, S.C.; Shukla, D. Allosteric control of a plant receptor kinase through S-glutathionylation. Biophys. J. 2017, 113, 2354–2363. [Google Scholar] [CrossRef]
- Zhou, L.; Chan, J.C.Y.; Chupin, S.; Gueguen, N.; Desquiret-Dumas, V.; Koh, S.K.; Li, J.; Gao, Y.; Deng, L.; Verma, C.; et al. Increased protein S-glutathionylation in Leber’s hereditary optic neuropathy (LHON). Int. J. Mol. Sci. 2020, 21, 3027. [Google Scholar] [CrossRef]
- Zaffagnini, M.; Marchand, C.H.; Malferrari, M.; Murail, S.; Bonacchi, S.; Genovese, D.; Montalti, M.; Venturoli, G.; Falini, G.; Baaden, M.; et al. Glutathionylation primes soluble glyceraldehyde-3-phosphate dehydrogenase for late collapse into insoluble aggregates. Proc. Natl. Acad. Sci. USA 2019, 116, 26057–26065. [Google Scholar] [CrossRef]
- Hameed, M.S. S-Glutathionylation of cysteine-217 allosterically inhibits triose-phosphate isomerase: A molecular dynamics study. J. Proteins Proteom. 2016, 7. [Google Scholar]
- Bao, R.; Zhang, Y.; Lou, X.; Zhou, C.Z.; Chen, Y. Structural and kinetic analysis of Saccharomyces cerevisiae thioredoxin Trx1: Implications for the catalytic mechanism of GSSG reduced by the thioredoxin system. Biochim. Biophys. Acta 2009, 1794, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Chibani, K.; Saul, F.; Didierjean, C.; Rouhier, N.; Haouz, A. Structural snapshots along the reaction mechanism of the atypical poplar thioredoxin-like2. 1. FEBS Lett. 2018, 592, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; San Koh, C.; Zaffagnini, M.; Winger, A.M.; Gualberto, J.M.; Corbier, C.; Decottignies, P.; Jacquot, J.P.; Lemaire, S.D.; Didierjean, C.; et al. Structure-function relationship of the chloroplastic glutaredoxin S12 with an atypical WCSYS active site. J. Biol. Chem. 2009, 284, 9299–9310. [Google Scholar] [CrossRef]
- Couturier, J.; Ströher, E.; Albetel, A.N.; Roret, T.; Muthuramalingam, M.; Tarrago, L.; Seidel, T.; Tsan, P.; Jacquot, J.P.; Johnson, M.K.; et al. Arabidopsis chloroplastic glutaredoxin C5 as a model to explore molecular determinants for iron-sulfur cluster binding into glutaredoxins. J. Biol. Chem. 2011, 286, 27515–27527. [Google Scholar] [CrossRef]
- Discola, K.F.; de Oliveira, M.A.; Cussiol, J.R.R.; Monteiro, G.; Bárcena, J.A.; Porras, P.; Padilla, C.A.; Guimarães, B.G.; Netto, L.E.S. Structural aspects of the distinct biochemical properties of glutaredoxin 1 and glutaredoxin 2 from Saccharomyces cerevisiae. J. Mol. Biol. 2009, 385, 889–901. [Google Scholar] [CrossRef]
- Lallement, P.A.; Meux, E.; Gualberto, J.M.; Prosper, P.; Didierjean, C.; Saul, F.; Haouz, A.; Rouhier, N.; Hecker, A. Structural and enzymatic insights into Lambda glutathione transferases from Populus trichocarpa, monomeric enzymes constituting an early divergent class specific to terrestrial plants. Biochem. J. 2014, 462, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Brock, J.; Liu, D.; Board, P.G.; Oakley, A.J. Structural insights into the dehydroascorbate reductase activity of human omega-class glutathione transferases. J. Mol. Biol. 2012, 420, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Perrot, T.; Schwartz, M.; Deroy, A.; Girardet, J.M.; Kohler, A.; Morel-Rouhier, M.; Favier, F.; Gelhaye, E.; Didierjean, C. Diversity of Omega Glutathione Transferases in mushroom-forming fungi revealed by phylogenetic, transcriptomic, biochemical and structural approaches. Fungal Genet. Biol. 2021, 148, 103506. [Google Scholar] [CrossRef] [PubMed]
- Pégeot, H.; Koh, C.S.; Petre, B.; Mathiot, S.; Duplessis, S.; Hecker, A.; Didierjean, C.; Rouhier, N. The poplar Phi class glutathione transferase: Expression, activity and structure of GSTF1. Front. Plant Sci. 2014, 5, 712. [Google Scholar] [PubMed]
- Inaka, K.; Miki, K.; Kikuchi, M.; Taniyama, Y.; Matsushima, M. Structure of a glutathionylated human lysozyme: A folding intermediate mimic in the formation of a disulfide bond. Acta Crystallogr. Sect. D Biol. Crystallogr. 1995, 51, 619–625. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 03; Gaussian Inc.: Wallingford, CT, USA, 2016; Volume 3. [Google Scholar]
- Case, D.; Belfon, K.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham III, T.; Cruzeiro, V.; Darden, T.; Duke, R.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins Struct. Funct. Genet. 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Z’idek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham III, T.E. Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using water-model-specific ion parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N · log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Organism | Abbreviation | PDB ID | Resolution (Å) | Oxidation Site |
|---|---|---|---|---|---|
| Thioredoxin 1 | Saccharomyces cerevisiae | ScTrx1 | 3F3R [26] | 1.80 | C30 |
| Thioredoxin-like2.1 | Populus tremula x tremuloides | PtTrxL2.1 | 5NYN [27] | 1.60 | C67 |
| Glutaredoxin S12 | Populus tremula x tremuloides | PtGrxS12 | 3FZ9 [28] | 1.70 | C29 |
| Glutaredoxin C5 | Aradobidopsis thaliana | AtGrxC5 | 3RHB [29] | 1.20 | C29 |
| Glutaredoxin 2 | Saccharomyces cerevisiaes | ScGrx2 | 3D5J [30] | 1.91 | C30 |
| Glutathione transferase 1 | Populus trichocarpa | PtGSTL1 | 4PQH [31] | 1.40 | C36 |
| Glutathione transferase 3 | Populus trichocarpa | PtGSTL3 | 4PQI [31] | 1.95 | C41 |
| Glutathione transferase 2 | Homo sapiens | hGSTO2 | 3Q19 1 [32] | 1.90 | C29 |
| Glutathione transferase 2C | Trametes versicolor | TvGSTO2C | 6SR8 1 [33] | 1.94 | C30 |
| Glutathione transferase F1 | Populus tremula x tremuloides | PtGSTF1 | 4RI7 1 [34] | 1.80 | C30 |
| Lysosyme | Homo sapiens | hLyso | 1HNL [35] | 1.40 | C95 |
| System | Reference Rg | AF Simulations Rg | crys Simulations Rg |
|---|---|---|---|
| AtGrxC5 | 13.2 | 13.3 ± 0.1 | 13.3 ± 0.1 |
| hLyso | 14.3 | 14.4 ± 0.1 | 14.4 ± 0.1 |
| hGSTO2 | 22.6 | 22.4 ± 0.2 | 22.7 ± 0.2 |
| PtGSTL1 | 16.9 | 17.2 ± 0.1 | 17.2 ± 0.1 |
| PtGSTL3 | 17.1 | 17.4 ± 0.4 | 17.4 ± 0.1 |
| PtGrxS12 | 13.2 | 13.4 ± 0.1 | 13.4 ± 0.1 |
| PtGSTF1 | 20.7 | 21.0 ± 0.1 | 21.0 ± 0.1 |
| PtTrxL2.1 | 13.5 | 13.7 ± 0.1 | 13.7 ± 0.1 |
| ScGrx2 | 13.0 | 13.4 ± 0.1 | 13.4 ± 0.1 |
| ScTrx1 | 12.4 | 12.8 ± 0.1 | 12.7 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elftmaoui, Z.; Bignon, E. Robust AMBER Force Field Parameters for Glutathionylated Cysteines. Int. J. Mol. Sci. 2023, 24, 15022. https://doi.org/10.3390/ijms241915022
Elftmaoui Z, Bignon E. Robust AMBER Force Field Parameters for Glutathionylated Cysteines. International Journal of Molecular Sciences. 2023; 24(19):15022. https://doi.org/10.3390/ijms241915022
Chicago/Turabian StyleElftmaoui, Zineb, and Emmanuelle Bignon. 2023. "Robust AMBER Force Field Parameters for Glutathionylated Cysteines" International Journal of Molecular Sciences 24, no. 19: 15022. https://doi.org/10.3390/ijms241915022