

Study of Ubiquitin Pathway Genes in a French Population with Amyotrophic Lateral Sclerosis: Focus on HECW1 Encoding the E3 Ligase NEDL1

 ,
,  , , , ,
, , , , 

Abstract
:1. Introduction
2. Results
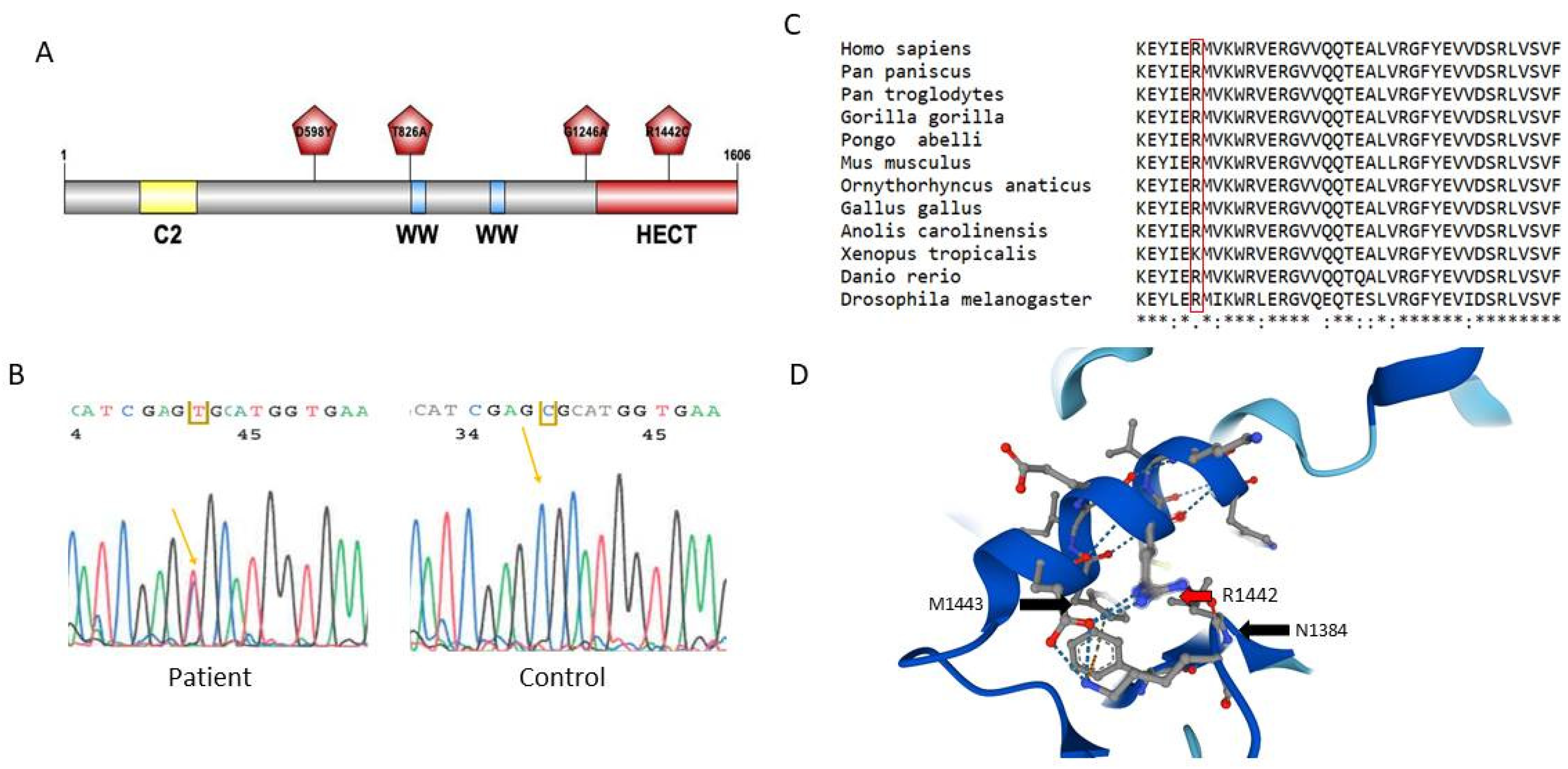
2.1. Genetic Screening of 12 Genes of the Ubiquitin Pathway in ALS
2.2. Genetic Screening on a Second Cohort of ALS Patients
2.3. Study of Neural Expression of NEDL1 Encoded by HECW1
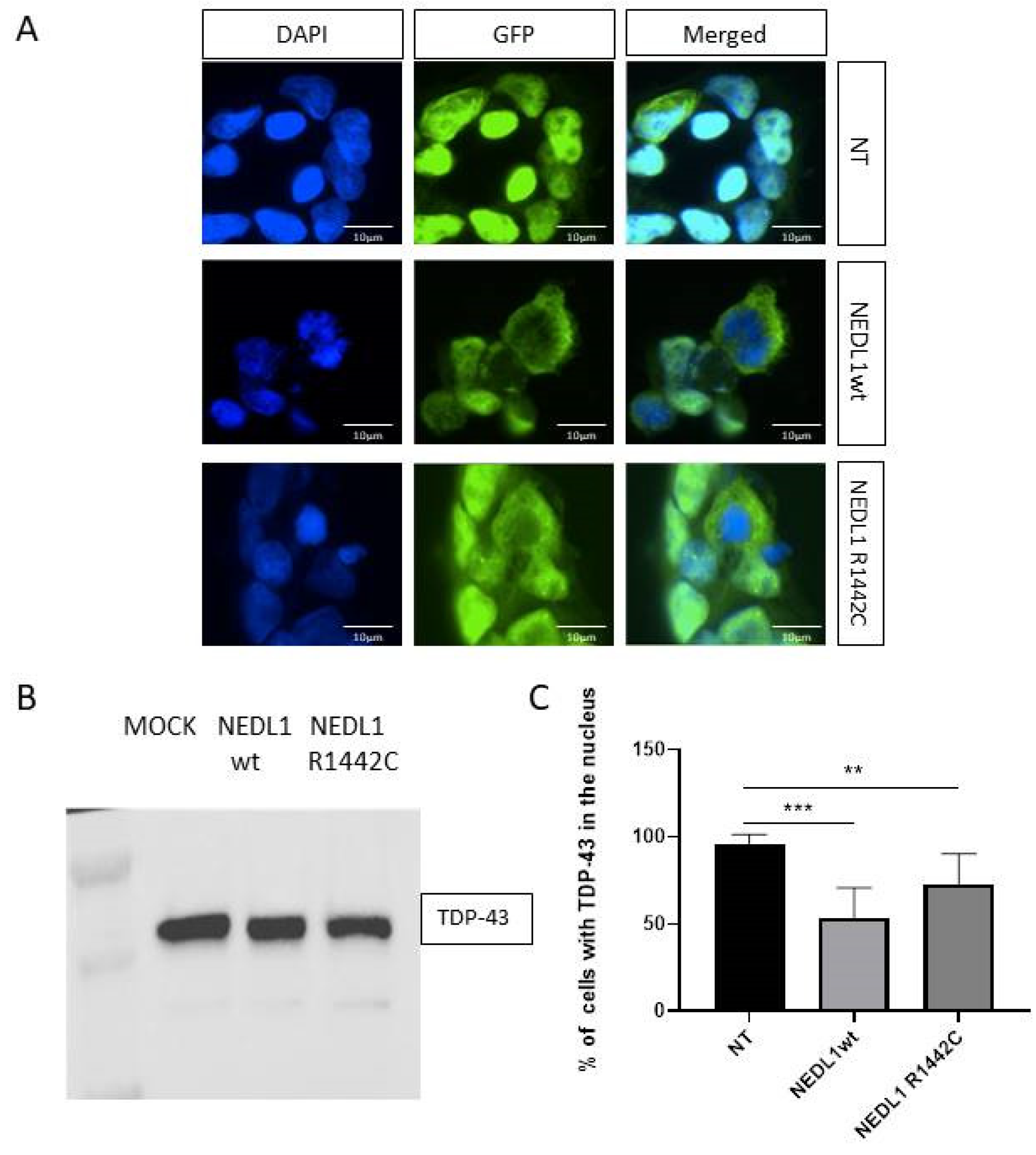
2.4. Functional Study of the NEDL1 Protein
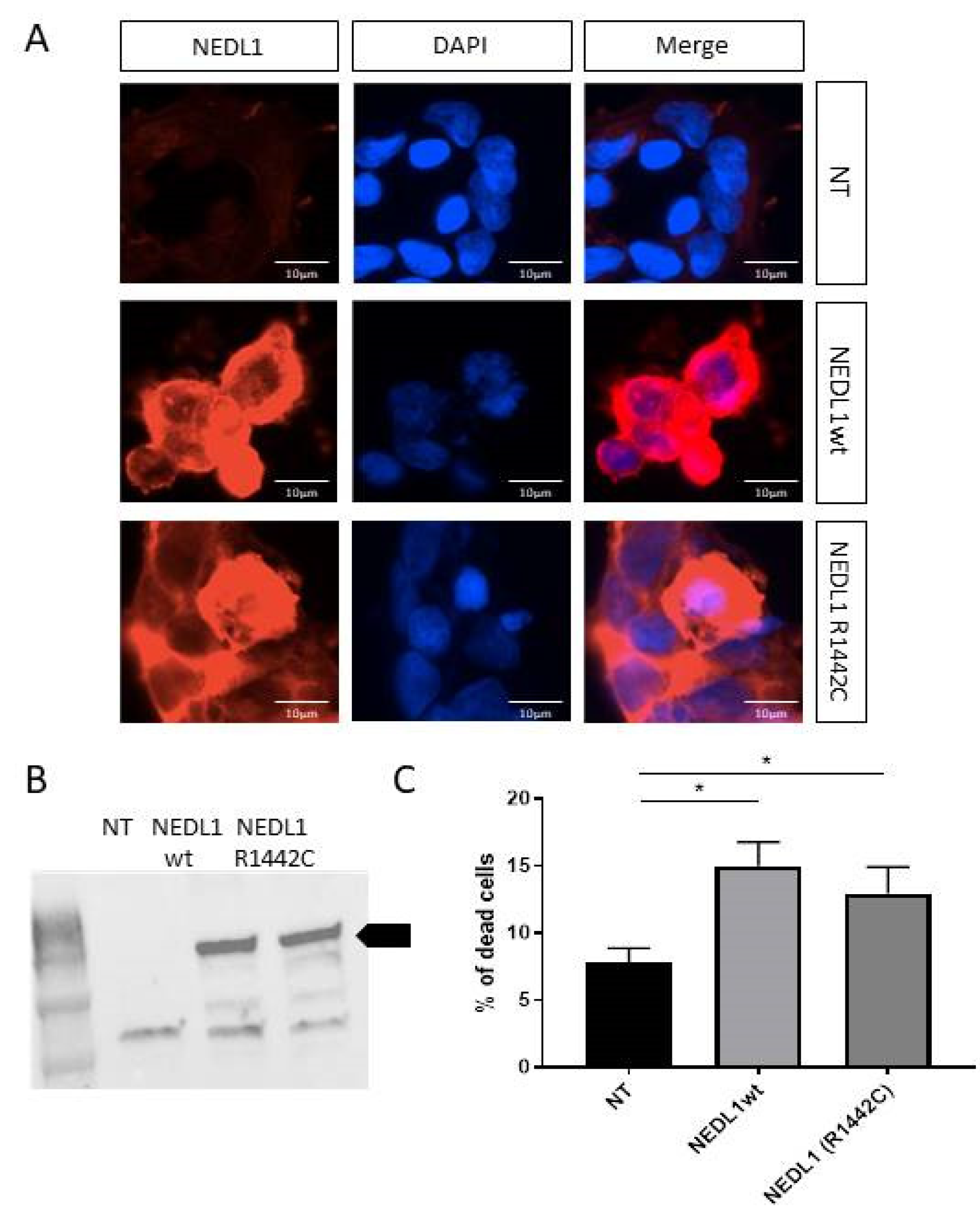
3. Discussion
4. Materials and Methods
4.1. Patients with ALS
4.2. Genetic Study by Next Generation Sequencing
4.3. Plasmid Constructs
4.4. Cell Cultures and Transfections
4.5. RNA Extraction
4.6. Gene Expression Studies
4.7. Cell Viability Assay
4.8. Immunocytochemistry
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.L.; Topp, S.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, A.; Joshi, V.; Amanullah, A.; Mishra, R.; Arora, N.; Prasad, A.; Mishra, A. E3 Ubiquitin Ligases Neurobiological Mechanisms: Development to Degeneration. Front. Mol. Neurosci. 2017, 10, 151. [Google Scholar] [CrossRef]
- Haouari, S.; Vourc’h, P.; Jeanne, M.; Marouillat, S.; Veyrat-Durebex, C.; Lanznaster, D.; Laumonnier, F.; Corcia, P.; Blasco, H.; Andres, C.R. The Roles of NEDD4 Subfamily of HECT E3 Ubiquitin Ligases in Neurodevelopment and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 3882. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Zhang, L.; Haraguchi, S.; Koda, T.; Hashimoto, K.; Nakagawara, A. Muscle atrophy and motor neuron degeneration in human NEDL1 transgenic mice. J. Biomed. Biotechnol. 2011, 2011, 831092. [Google Scholar] [CrossRef]
- Dangoumau, A.; Marouillat, S.; Coelho, R.; Wurmser, F.; Brulard, C.; Haouari, S.; Laumonnier, F.; Corcia, P.; Andres, C.R.; Blasco, H.; et al. Dysregulations of Expression of Genes of the Ubiquitin/SUMO Pathways in an In Vitro Model of Amyotrophic Lateral Sclerosis Combining Oxidative Stress and SOD1 Gene Mutation. Int. J. Mol. Sci. 2021, 22, 1796. [Google Scholar] [CrossRef] [PubMed]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.-P. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Camu, W.; Brulard, C.; Marouillat, S.; Couratier, P.; Camdessanché, J.-P.; Cintas, P.; Verschueren, A.; Soriani, M.-H.; Desnuelle, C.; et al. Effect of familial clustering in the genetic screening of 235 French ALS families. J. Neurol. Neurosurg. Psychiatry 2021, 92, 479–484. [Google Scholar] [CrossRef]
- Oh, S.-M.; Liu, Z.; Okada, M.; Jang, S.-W.; Liu, X.; Chan, C.-B.; Luo, H.; Ye, K. Ebp1 sumoylation, regulated by TLS/FUS E3 ligase, is required for its anti-proliferative activity. Oncogene 2010, 29, 1017–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, I.P.; Williams, K.L.; Warraich, S.T.; Durnall, J.C.; Thoeng, A.D.; Manavis, J.; Blumbergs, P.C.; Vucic, S.; Kiernan, M.C.; Nicholson, G.A. FUS mutations in amyotrophic lateral sclerosis: Clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, F.; Hu, Y.; Chen, R.; Meng, D.; Guo, L.; Lv, H.; Guan, J.; Jia, Y. In vivo stress granule misprocessing evidenced in a FUS knock-in ALS mouse model. Brain 2020, 143, 1350–1367. [Google Scholar] [CrossRef]
- D’Angiolella, V.; Donato, V.; Vijayakumar, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Dynlacht, B.; Pagano, M. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 2010, 466, 138–142. [Google Scholar] [CrossRef]
- Miyazaki, K.; Fujita, T.; Ozaki, T.; Kato, C.; Kurose, Y.; Sakamoto, M.; Kato, S.; Goto, T.; Itoyama, Y.; Aoki, M.; et al. NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J. Biol. Chem. 2004, 279, 11327–11335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Fukuda, Y.; Yoshimura, J.; Toyoda, A.; Kurppa, K.; Moritoyo, H.; Belzil, V.V.; Dion, P.A.; Higasa, K.; Doi, K.; et al. ERBB4 Mutations that Disrupt the Neuregulin-ErbB4 Pathway Cause Amyotrophic Lateral Sclerosis Type 19. Am. J. Hum. Genet. 2013, 93, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, Z.; Alimandi, M.; Chen, C. WW domain containing E3 ubiquitin protein ligase 1 targets the full-length ErbB4 for ubiquitin-mediated degradation in breast cancer. Oncogene 2009, 28, 2948–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Liu, H.; Xu, C. Estrogen degrades Scribble in endometrial epithelial cells through E3 ubiquitin ligase HECW1 in the development of diffuse adenomyosis. Biol. Reprod. 2020, 102, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dong, S.; Li, L.; Wang, H.; Zhao, J.; Zhao, Y. The E3 ubiquitin ligase HECW1 targets thyroid transcription factor 1 (TTF1/NKX2.1) for its degradation in the ubiquitin-proteasome system. Cell Signal. 2019, 58, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ning, G.; Si, P.; Zhang, C.; Liu, W.; Ge, W.; Cui, K.; Zhang, R.; Ge, S. E3 ubiquitin ligase HECW1 promotes the metastasis of non-small cell lung cancer cells through mediating the ubiquitination of Smad4. Biochem. Cell Biol. 2021, 99, 675–681. [Google Scholar] [CrossRef]
- Plant, P.J.; Yeger, H.; Staub, O.; Howard, P.; Rotin, D. The C2 domain of the ubiquitin protein ligase Nedd4 mediates Ca2+-dependent plasma membrane localization. J. Biol. Chem. 1997, 272, 32329–32336. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ozaki, T.; Kikuchi, H.; Yamamoto, H.; Ohira, M.; Nakagawara, A. A novel HECT-type E3 ubiquitin protein ligase NEDL1 enhances the p53-mediated apoptotic cell death in its catalytic activity-independent manner. Oncogene 2008, 27, 3700–3709. [Google Scholar] [CrossRef] [Green Version]
- Maor-Nof, M.; Shipony, Z.; Lopez-Gonzalez, R.; Nakayama, L.; Zhang, Y.-J.; Couthouis, J.; Blum, J.A.; Castruita, P.A.; Linares, G.R.; Ruan, K.; et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 2021, 184, 689–708.e20. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.P.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picher-Martel, V.; Dutta, K.; Phaneuf, D.; Sobue, G.; Julien, J.-P. Ubiquilin-2 drives NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol. Brain 2015, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hans, F.; Eckert, M.; von Zweydorf, F.; Gloeckner, C.J.; Kahle, P.J. Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2018, 293, 16083–16099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Nonoka, T.; Kametani, F.; Yamashita, M.; Hosokawa, M.; Niizato, K.; Tsuchiya, K.; Kobayashi, Z.; Ikeda, K.; et al. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology 2010, 30, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Prater, K.E.; Latimer, C.S.; Jayadev, S. Glial TDP-43 and TDP-43 induced glial pathology, focus on neurodegenerative proteinopathy syndromes. Glia 2022, 70, 239–255. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Locations | Proteins | Functions | Variants in the Present Study | Cohort Number | Freq. MinE ALS Cases # | Freq. MinE Controls # | Freq. gnomAD | Condition in ClinVar | Class (ACMG) |
|---|---|---|---|---|---|---|---|---|---|---|
| CCNF | 16p13.3 | Cyclin F | Element of E3 ligase complex | S3G | 2 | 0 | 0 | 0.00006 | NR | 2 |
| A74T | 2 | 0.00015 | 0 | 0.00006 | NR | 2 | ||||
| R406Q | 2 | 0.00255 | LB | 2 | ||||||
| R521H | 2 | 0 | 0 | 0.00001 | NR | 3 | ||||
| P549S | 2 | 0 | 0 | 0.00001 | NR | 3 | ||||
| S621G | 2 | 0 | 0 | 0.00042 | P | 5 | ||||
| FBXO32 | 8q24.13 | F-box protein 32 | E3 ligase | * | 1 | |||||
| FUS | 16p11.2 | FUS | E3 ligase, RBP | H517T | 2 | 0 | 0 | 0 | NR | 4 |
| R518K | 1 | 0 | 0 | 0 | NR | 3 | ||||
| R521S | 1 | 0 | 0 | 0 | NR | 4 | ||||
| R521C | 1 | 0 | 0 | 0.00001 | P | 5 | ||||
| HECW1 | 7p14.1-p13 | NEDL1 | E3 ligase | V196I | 1 | 0 | 0.00546 | 0.00003 | NR | 3 |
| E502Q | 1 | 0.00515 | 0.00300 | 0.00223 | NR | 2 | ||||
| G576D | 1 | 0.00046 | 0.00055 | 0.00061 | NR | 2 | ||||
| D598Y | 2 | 0 | 0 | 0 | NR | 3 | ||||
| E820G | 2 | 0 | 0 | 0.00268 | NR | 2 | ||||
| T826A | 1 | 0 | 0 | 0 | NR | 3 | ||||
| D1005E | 1 | 0.00241 | 0.00164 | 0.00184 | NR | 2 | ||||
| V1099L | 2 | 0.00252 | 0.00328 | 0.00061 | NR | 3 | ||||
| V1184T | 2 | 0 | 0 | 0 | NR | 2 | ||||
| G1246A | 1 | 0 | 0 | 0 | NR | 3 | ||||
| R1442C | 1 | 0 | 0 | 0 | NR | 3 | ||||
| KDM2B | 12q24.31 | Lysine demethylase 2B | Subunit CUL1-RING E3 ligase | H19R | 1 | 0.00733 | 0.00655 | 0.00714 | NR | 2 |
| T28fs | 1 | 0.00470 | 0.00300 | 0.00304 | NR | 2 | ||||
| MARCH5 | 10q23.32-33 | RNF135 | E3 ligase | * | 1 | |||||
| RBX1 | 22q13.2 | Ring Box 1 | E3 ligase | * | 1 | |||||
| RNF19A | 8q22.2 | Ring finger protein 19A | E3 ligase | A784T | 1 | 0.00115 | 0 | 0.00007 | NR | 2 |
| TRIM63 | 1p36.11 | TRIM63 | E3 ligase | A48V | 1 | 0.00241 | 0.00300 | 0.00808 | NR | 2 |
| UBE2D2 | 5q31.2 | UBE2D2 | E2 conjugating enzyme | * | 1 | |||||
| UBE2D3 | 4q24 | UBE2D3 | E2 conjugating enzyme | * | 1 | |||||
| UBQLN2 | Xp11.21 | Ubiquilin 2 | Shuttle protein in the Ub system | S371N | 2 | 0 | 0 | 0 | NR | 3 |
| UHRF2 | 9p24.1 | UHRF2 | E3 ligase | * | 1 | |||||
| VCP | 9p13.3 | Valosin containing protein | Binds ubiquitynated substrates | * | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haouari, S.; Andres, C.R.; Lanznaster, D.; Marouillat, S.; Brulard, C.; Dangoumau, A.; Ung, D.; Veyrat-Durebex, C.; Laumonnier, F.; Blasco, H.; et al. Study of Ubiquitin Pathway Genes in a French Population with Amyotrophic Lateral Sclerosis: Focus on HECW1 Encoding the E3 Ligase NEDL1. Int. J. Mol. Sci. 2023, 24, 1268. https://doi.org/10.3390/ijms24021268
Haouari S, Andres CR, Lanznaster D, Marouillat S, Brulard C, Dangoumau A, Ung D, Veyrat-Durebex C, Laumonnier F, Blasco H, et al. Study of Ubiquitin Pathway Genes in a French Population with Amyotrophic Lateral Sclerosis: Focus on HECW1 Encoding the E3 Ligase NEDL1. International Journal of Molecular Sciences. 2023; 24(2):1268. https://doi.org/10.3390/ijms24021268
Chicago/Turabian StyleHaouari, Shanez, Christian Robert Andres, Debora Lanznaster, Sylviane Marouillat, Céline Brulard, Audrey Dangoumau, Devina Ung, Charlotte Veyrat-Durebex, Frédéric Laumonnier, Hélène Blasco, and et al. 2023. "Study of Ubiquitin Pathway Genes in a French Population with Amyotrophic Lateral Sclerosis: Focus on HECW1 Encoding the E3 Ligase NEDL1" International Journal of Molecular Sciences 24, no. 2: 1268. https://doi.org/10.3390/ijms24021268