
Regulation of Oocyte Apoptosis: A View from Gene Knockout Mice



Abstract
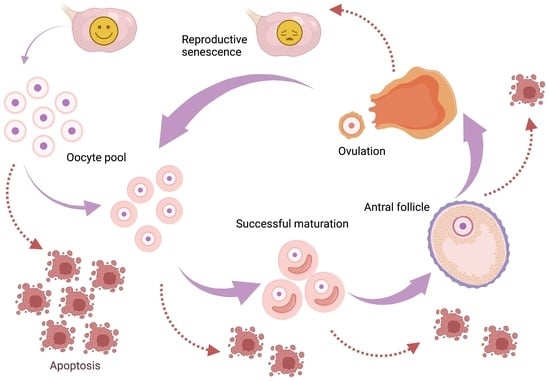
1. Introduction
2. Apoptosis Pathways
3. Knocking Out Pro-Survival Bcl-2 Family Genes
4. Knocking Out Pro-Apoptotic Bcl-2 Family Genes
5. Caspase Knockout Mice
6. The Extrinsic Pathway
7. p53 Family Proteins
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forabosco, A.; Sforza, C.; Pol, A.D.; Vizzotto, L.; Marzona, L.; Ferrario, V.F. Morphometric Study of the Human Neonatal Ovary. Anat. Rec. 1991, 231, 201–208. [Google Scholar] [CrossRef]
- Baker, T.G. A Quantitative and Cytological Study of Germ Cells in Human Ovaries. Proc. Royal Soc. Lond. Ser. B Biol. Sci. 1963, 158, 417–433. [Google Scholar] [CrossRef]
- Pepling, M.E.; Spradling, A.C. Mouse Ovarian Germ Cell Cysts Undergo Programmed Breakdown to Form Primordial Follicles. Dev. Biol. 2001, 234, 339–351. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Kurokawa, M.; Kornbluth, S. Caspases and Kinases in a Death Grip. Cell 2009, 138, 838–854. [Google Scholar] [CrossRef]
- Bao, Q.; Shi, Y. Apoptosome: A Platform for the Activation of Initiator Caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef]
- Brown-Suedel, A.N.; Bouchier-Hayes, L. Caspase-2 Substrates: To Apoptosis, Cell Cycle Control, and Beyond. Front. Cell Dev. Biol. 2020, 8, 610022. [Google Scholar] [CrossRef]
- Ratts, V.S.; Flaws, J.A.; Kolp, R.; Sorenson, C.M.; Tilly, J.L. Ablation of Bcl-2 Gene Expression Decreases the Numbers of Oocytes and Primordial Follicles Established in the Post-Natal Female Mouse Gonad. Endocrinology 1995, 136, 3665–3668. [Google Scholar] [CrossRef]
- Flaws, J.A.; Hirshfield, A.N.; Hewitt, J.A.; Babus, J.K.; Furth, P.A. Effect of Bcl-2 on the Primordial Follicle Endowment in the Mouse Ovary. Biol. Reprod. 2001, 64, 1153–1159. [Google Scholar] [CrossRef]
- Morita, Y.; Perez, G.I.; Maravei, D.V.; Tilly, K.I.; Tilly, J.L. Targeted Expression of Bcl-2 in Mouse Oocytes Inhibits Ovarian Follicle Atresia and Prevents Spontaneous and Chemotherapy-Induced Oocyte Apoptosis In Vitro. Mol. Endocrinol. Baltim. Md. 1999, 13, 841–850. [Google Scholar] [CrossRef]
- Jones, R.L.; Pepling, M.E. Role of the Antiapoptotic Proteins BCL2 and MCL1 in the Neonatal Mouse Ovary. Biol. Reprod. 2013, 88, 46. [Google Scholar] [CrossRef]
- Rucker, E.B.; Dierisseau, P.; Wagner, K.-U.; Garrett, L.; Wynshaw-Boris, A.; Flaws, J.A.; Hennighausen, L. Bcl-x and Bax Regulate Mouse Primordial Germ Cell Survival and Apoptosis during Embryogenesis. Mol. Endocrinol. 2000, 14, 1038–1052. [Google Scholar] [CrossRef]
- Riedlinger, G.; Okagaki, R.; Wagner, K.-U.; Rucker, E.B.; Oka, T.; Miyoshi, K.; Flaws, J.A.; Hennighausen, L. Bcl-x Is Not Required for Maintenance of Follicles and Corpus Luteum in the Postnatal Mouse Ovary. Biol. Reprod. 2002, 66, 438–444. [Google Scholar] [CrossRef]
- Omari, S.; Waters, M.; Naranian, T.; Kim, K.; Perumalsamy, A.L.; Chi, M.; Greenblatt, E.; Moley, K.H.; Opferman, J.T.; Jurisicova, A. Mcl-1 Is a Key Regulator of the Ovarian Reserve. Cell Death Dis. 2015, 6, e1755. [Google Scholar] [CrossRef]
- Print, C.G.; Loveland, K.L.; Gibson, L.; Meehan, T.; Stylianou, A.; Wreford, N.; de Kretser, D.; Metcalf, D.; Köntgen, F.; Adams, J.M.; et al. Apoptosis Regulator Bcl-w Is Essential for Spermatogenesis but Appears Otherwise Redundant. Proc. Natl. Acad. Sci. USA 1998, 95, 12424–12431. [Google Scholar] [CrossRef]
- Russell, H.R.; Lee, Y.; Miller, H.L.; Zhao, J.; McKinnon, P.J. Murine Ovarian Development Is Not Affected by Inactivation of the Bcl-2 Family Member Diva. Mol. Cell. Biol. 2002, 22, 6866–6870. [Google Scholar] [CrossRef]
- Schenk, R.L.; Tuzlak, S.; Carrington, E.M.; Zhan, Y.; Heinzel, S.; Teh, C.E.; Gray, D.H.; Tai, L.; Lew, A.M.; Villunger, A.; et al. Characterisation of Mice Lacking All Functional Isoforms of the Pro-Survival BCL-2 Family Member A1 Reveals Minor Defects in the Haematopoietic Compartment. Cell Death Differ. 2017, 24, 534–545. [Google Scholar] [CrossRef]
- Ranger, A.M.; Zha, J.; Harada, H.; Datta, S.R.; Danial, N.N.; Gilmore, A.P.; Kutok, J.L.; Beau, M.M.L.; Greenberg, M.E.; Korsmeyer, S.J. Bad-Deficient Mice Develop Diffuse Large B Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9324–9329. [Google Scholar] [CrossRef]
- Coultas, L.; Bouillet, P.; Stanley, E.G.; Brodnicki, T.C.; Adams, J.M.; Strasser, A. Proapoptotic BH3-Only Bcl-2 Family Member Bik/Blk/Nbk Is Expressed in Hemopoietic and Endothelial Cells but Is Redundant for Their Programmed Death. Mol. Cell Biol. 2004, 24, 1570–1581. [Google Scholar] [CrossRef]
- Kaufmann, T.; Tai, L.; Ekert, P.G.; Huang, D.C.S.; Norris, F.; Lindemann, R.K.; Johnstone, R.W.; Dixit, V.M.; Strasser, A. The BH3-Only Protein Bid Is Dispensable for DNA Damage- and Replicative Stress-Induced Apoptosis or Cell-Cycle Arrest. Cell 2007, 129, 423–433. [Google Scholar] [CrossRef]
- Yin, X.-M.; Wang, K.; Gross, A.; Zhao, Y.; Zinkel, S.; Klocke, B.; Roth, K.A.; Korsmeyer, S.J. Bid-Deficient Mice Are Resistant to Fas-Induced Hepatocellular Apoptosis. Nature 1999, 400, 886–891. [Google Scholar] [CrossRef]
- Kerr, J.B.; Hutt, K.J.; Michalak, E.M.; Cook, M.; Vandenberg, C.J.; Liew, S.H.; Bouillet, P.; Mills, A.; Scott, C.L.; Findlay, J.K.; et al. DNA Damage-Induced Primordial Follicle Oocyte Apoptosis and Loss of Fertility Require TAp63-Mediated Induction of Puma and Noxa. Mol. Cell 2012, 48, 343–352. [Google Scholar] [CrossRef]
- ElInati, E.; Zielinska, A.P.; McCarthy, A.; Kubikova, N.; Maciulyte, V.; Mahadevaiah, S.; Sangrithi, M.N.; Ojarikre, O.; Wells, D.; Niakan, K.K.; et al. The BCL-2 Pathway Preserves Mammalian Genome Integrity by Eliminating Recombination-Defective Oocytes. Nat. Commun. 2020, 11, 2598. [Google Scholar] [CrossRef]
- Coultas, L.; Bouillet, P.; Loveland, K.L.; Meachem, S.; Perlman, H.; Adams, J.M.; Strasser, A. Concomitant Loss of Proapoptotic BH3-only Bcl-2 Antagonists Bik and Bim Arrests Spermatogenesis. Embo J. 2005, 24, 3963–3973. [Google Scholar] [CrossRef]
- Kelly, P.N.; White, M.J.; Goschnick, M.W.; Fairfax, K.A.; Tarlinton, D.M.; Kinkel, S.A.; Bouillet, P.; Adams, J.M.; Kile, B.T.; Strasser, A. Individual and Overlapping Roles of BH3-Only Proteins Bim and Bad in Apoptosis of Lymphocytes and Platelets and in Suppression of Thymic Lymphoma Development. Cell Death Differ. 2010, 17, 1655–1664. [Google Scholar] [CrossRef]
- Ren, D.; Tu, H.-C.; Kim, H.; Wang, G.X.; Bean, G.R.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. BID, BIM, and PUMA Are Essential for Activation of the BAX- and BAK-Dependent Cell Death Program. Science 2010, 330, 1390–1393. [Google Scholar] [CrossRef]
- Liew, S.H.; Vaithiyanathan, K.; Cook, M.; Bouillet, P.; Scott, C.L.; Kerr, J.B.; Strasser, A.; Findlay, J.K.; Hutt, K.J. Loss of the Proapoptotic BH3-Only Protein BCL-2 Modifying Factor Prolongs the Fertile Life Span in Female Mice. Biol. Reprod. 2014, 90, 77. [Google Scholar] [CrossRef] [PubMed]
- Labi, V.; Erlacher, M.; Kiessling, S.; Manzl, C.; Frenzel, A.; O’Reilly, L.; Strasser, A.; Villunger, A. Loss of the BH3-Only Protein Bmf Impairs B Cell Homeostasis and Accelerates γ Irradiation–Induced Thymic Lymphoma Development. J. Exp. Med. 2008, 205, 641–655. [Google Scholar] [CrossRef]
- Coultas, L.; Terzano, S.; Thomas, T.; Voss, A.; Reid, K.; Stanley, E.G.; Scott, C.L.; Bouillet, P.; Bartlett, P.; Ham, J.; et al. Hrk/DP5 Contributes to the Apoptosis of Select Neuronal Populations but Is Dispensable for Haematopoietic Cell Apoptosis. J. Cell Sci. 2007, 120, 2044–2052. [Google Scholar] [CrossRef]
- Lindsten, T.; Ross, A.J.; King, A.; Zong, W.-X.; Rathmell, J.C.; Shiels, H.A.; Ulrich, E.; Waymire, K.G.; Mahar, P.; Frauwirth, K.; et al. The Combined Functions of Proapoptotic Bcl-2 Family Members Bak and Bax Are Essential for Normal Development of Multiple Tissues. Mol. Cell. 2000, 6, 1389–1399. [Google Scholar] [CrossRef]
- Ke, F.; Bouillet, P.; Kaufmann, T.; Strasser, A.; Kerr, J.; Voss, A.K. Consequences of the Combined Loss of BOK and BAK or BOK and BAX. Cell Death Dis. 2013, 4, e650. [Google Scholar] [CrossRef]
- Perez, G.I.; Robles, R.; Knudson, C.M.; Flaws, J.A.; Korsmeyer, S.J.; Tilly, J.L. Prolongation of Ovarian Lifespan into Advanced Chronological Age by Bax-Deficiency. Nat. Genet. 1999, 21, 200–203. [Google Scholar] [CrossRef]
- Ke, F.; Voss, A.; Kerr, J.B.; O’Reilly, L.A.; Tai, L.; Echeverry, N.; Bouillet, P.; Strasser, A.; Kaufmann, T. BCL-2 Family Member BOK Is Widely Expressed but Its Loss Has Only Minimal Impact in Mice. Cell Death Differ. 2012, 19, 915–925. [Google Scholar] [CrossRef]
- Matikainen, T.; Perez, G.I.; Zheng, T.S.; Kluzak, T.R.; Rueda, B.R.; Flavell, R.A.; Tilly, J.L. Caspase-3 Gene Knockout Defines Cell Lineage Specificity for Programmed Cell Death Signaling in the Ovary. Endocrinology 2001, 142, 2468–2480. [Google Scholar] [CrossRef]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key Mediators of Mitochondrial Events of Apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef]
- Bergeron, L.; Perez, G.I.; Macdonald, G.; Shi, L.; Sun, Y.; Jurisicova, A.; Varmuza, S.; Latham, K.E.; Flaws, J.A.; Salter, J.C.M.; et al. Defects in Regulation of Apoptosis in Caspase-2-Deficient Mice. Gene Dev. 1998, 12, 1304–1314. [Google Scholar] [CrossRef]
- Morita, Y.; Maravei, D.V.; Bergeron, L.; Wang, S.; Perez, G.I.; Tsutsumi, O.; Taketani, Y.; Asano, M.; Horai, R.; Korsmeyer, S.J.; et al. Caspase-2 Deficiency Prevents Programmed Germ Cell Death Resulting from Cytokine Insufficiency but Not Meiotic Defects Caused by Loss of Ataxia Telangiectasia-Mutated (Atm) Gene Function. Cell Death Differ. 2001, 8, 614–620. [Google Scholar] [CrossRef]
- Ene, A.C.; Park, S.; Edelmann, W.; Taketo, T. Caspase 9 Is Constitutively Activated in Mouse Oocytes and Plays a Key Role in Oocyte Elimination during Meiotic Prophase Progression. Dev. Biol. 2013, 377, 213. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, K.; Yoshida, H.; Nishimura, Y.; Nishikawa, S.; Manabe, N.; Yonehara, S. Involvement of Fas Antigen in Ovarian Follicular Atresia and Luteolysis. Mol. Reprod. Dev. 1997, 47, 11–18. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Sakamaki, K.; Akazawa, Y.; Miyano, T. Oocyte Growth and Follicular Development in KIT-Deficient Fas-Knockout Mice. Reproduction 2007, 133, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Yang, G.; Pan, J.; Zhang, C. Tumor Necrosis Factor α Knockout Increases Fertility of Mice. Theriogenology 2010, 75, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Greenfeld, C.R.; Roby, K.F.; Pepling, M.E.; Babus, J.K.; Terranova, P.F.; Flaws, J.A. Tumor Necrosis Factor (TNF) Receptor Type 2 Is an Important Mediator of TNF Alpha Function in the Mouse Ovary. Biol. Reprod. 2007, 76, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, N.; Wang, F.; Roth, K.A.; Sawa, H.; Nakayama, K.; Nakayama, K.; Negishi, I.; Senju, S.; Zhang, Q.; Fujii, S.; et al. Massive Cell Death of Immature Hematopoietic Cells and Neurons in Bcl-x-Deficient Mice. Science 1995, 267, 1506–1510. [Google Scholar] [CrossRef]
- Rinkenberger, J.L.; Horning, S.; Klocke, B.; Roth, K.; Korsmeyer, S.J. Mcl-1 Deficiency Results in Peri-Implantation Embryonic Lethality. Gene Dev. 2000, 14, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Knudson, C.M.; Tung, K.S.K.; Tourtellotte, W.G.; Brown, G.A.J.; Korsmeyer, S.J. Bax-Deficient Mice with Lymphoid Hyperplasia and Male Germ Cell Death. Science 1995, 270, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Lindsten, T.; Zong, W.-X.; Cinalli, R.M.; Thompson, C.B. Deficiency in Bak and Bax Perturbs Thymic Selection and Lymphoid Homeostasis. Nat. Immunol. 2002, 3, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.Y.; Kaipia, A.; McGee, E.; Lomeli, M.; Hsueh, A.J.W. Bok Is a Pro-Apoptotic Bcl-2 Protein with Restricted Expression in Reproductive Tissues and Heterodimerizes with Selective Anti-Apoptotic Bcl-2 Family Members. Proc. Natl. Acad. Sci. USA 1997, 94, 12401–12406. [Google Scholar] [CrossRef]
- Jääskeläinen, M.; Nieminen, A.; Pökkylä, R.-M.; Kauppinen, M.; Liakka, A.; Heikinheimo, M.; Vaskivuo, T.E.; Klefström, J.; Tapanainen, J.S. Regulation of Cell Death in Human Fetal and Adult Ovaries—Role of Bok and Bcl-XL. Mol. Cell Endocrinol. 2010, 330, 17–24. [Google Scholar] [CrossRef]
- Takai, Y.; Matikainen, T.; Jurisicova, A.; Kim, M.R.; Trbovich, A.M.; Fujita, E.; Nakagawa, T.; Lemmers, B.; Flavell, R.A.; Hakem, R.; et al. Caspase-12 Compensates for Lack of Caspase-2 and Caspase-3 in Female Germ Cells. Apoptosis 2007, 12, 791–800. [Google Scholar] [CrossRef]
- Naz, R.; Zhu, X.; Menge, A. Expression of Tumor Necrosis Factor-a and Its Receptors Type I and Type II in Human Oocytes. Mol. Reprod. Dev. 1997, 47, 127–133. [Google Scholar] [CrossRef]
- Terranova, P.F. Potential Roles of Tumor Necrosis Factor-α in Follicular Development, Ovulation, and the Life Span of the Corpus Luteum. Domest. Anim. Endocrin. 1997, 14, 1–15. [Google Scholar] [CrossRef]
- Bang, S.; Kaur, S.; Kurokawa, M. Regulation of the p53 Family Proteins by the Ubiquitin Proteasomal Pathway. Int. J. Mol. Sci. 2019, 21, 261. [Google Scholar] [CrossRef] [PubMed]
- Viganò, M.A.; Mantovani, R. Hitting the Numbers: The Emerging Network of p63 Targets. Cell Cycle 2007, 6, 233–239. [Google Scholar] [CrossRef]
- Pyati, U.J.; Gjini, E.; Carbonneau, S.; Lee, J.-S.; Guo, F.; Jette, C.A.; Kelsell, D.P.; Look, A.T. p63 Mediates an Apoptotic Response to Pharmacological and Disease-Related ER Stress in the Developing Epidermis. Dev. Cell 2010, 21, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.-P.P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour Suppression by p53: A Role for the DNA Damage Response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef]
- Livera, G.; Uzbekov, R.; Jarrier, P.; Fouchécourt, S.; Duquenne, C.; Parent, A.; Marine, J.; Monget, P. Loss of Oocytes Due to Conditional Ablation of Murine Double Minute 2 (Mdm2) Gene Is p53-dependent and Results in Female Sterility. FEBS Lett. 2016, 590, 2566–2574. [Google Scholar] [CrossRef]
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule Is a Critical Mediator of the ARF Tumor Suppressor. Cell 2005, 121, 1071–1083. [Google Scholar] [CrossRef]
- Eisa, A.A.; Bang, S.; Crawford, K.J.; Murphy, E.M.; Feng, W.W.; Dey, S.; Wells, W.; Kon, N.; Gu, W.; Mehlmann, L.M.; et al. X-Linked Huwe1 Is Essential for Oocyte Maturation and Preimplantation Embryo Development. Iscience 2020, 23, 101523. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 Homolog at 3q27–29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Trink, B.; Okami, K.; Wu, L.; Sriuranpong, V.; Jen, J.; Sidransky, D. A New Human p53 Homologue. Nat. Med. 1998, 4, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Ohba, M.; Kawahara, C.; Ishioka, C.; Kanamaru, R.; Katoh, I.; Ikawa, Y.; Nimura, Y.; Nakagawara, A.; Obinata, M.; et al. Cloning and Functional Analysis of Human P51, Which Structurally and Functionally Resembles p53. Nat. Med. 1998, 4, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Kaelin, W.G. P53 Family Update: p73 and p63 Develop Their Own Identities. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 2001, 12, 337–349. [Google Scholar]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 Is Essential for Regenerative Proliferation in Limb, Craniofacial and Epithelial Development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Mills, A.A.; Zheng, B.; Wang, X.-J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 Is a p53 Homologue Required for Limb and Epidermal Morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef]
- Suh, E.-K.; Yang, A.; Kettenbach, A.; Bamberger, C.; Michaelis, A.H.; Zhu, Z.; Elvin, J.A.; Bronson, R.T.; Crum, C.P.; McKeon, F. p63 Protects the Female Germ Line during Meiotic Arrest. Nature 2006, 444, 624–628. [Google Scholar] [CrossRef]
- Gonfloni, S.; Tella, L.D.; Caldarola, S.; Cannata, S.M.; Klinger, F.G.; Bartolomeo, C.D.; Mattei, M.; Candi, E.; Felici, M.D.; Melino, G.; et al. Inhibition of the C-Abl–TAp63 Pathway Protects Mouse Oocytes from Chemotherapy-Induced Death. Nat. Med. 2009, 15, 1179–1185. [Google Scholar] [CrossRef]
- Bolcun-Filas, E.; Rinaldi, V.D.; White, M.E.; Schimenti, J.C. Reversal of Female Infertility by Chk2 Ablation Reveals the Oocyte DNA Damage Checkpoint Pathway. Science 2014, 343, 533–536. [Google Scholar] [CrossRef]
- Ruth, K.S.; Day, F.R.; Hussain, J.; Martínez-Marchal, A.; Aiken, C.E.; Azad, A.; Thompson, D.J.; Knoblochova, L.; Abe, H.; Tarry-Adkins, J.L.; et al. Genetic Insights into Biological Mechanisms Governing Human Ovarian Ageing. Nature 2021, 596, 393–397. [Google Scholar] [CrossRef]
- Xu, Y.; Ashley, T.; Brainerd, E.E.; Bronson, R.T.; Meyn, M.S.; Baltimore, D. Targeted Disruption of ATM Leads to Growth Retardation, Chromosomal Fragmentation during Meiosis, Immune Defects, and Thymic Lymphoma. Gene Dev. 1996, 10, 2411–2422. [Google Scholar] [CrossRef]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-Deficient Mice: A Paradigm of Ataxia Telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef]
- Brown, E.J.; Baltimore, D. ATR Disruption Leads to Chromosomal Fragmentation and Early Embryonic Lethality. Gene Dev. 2000, 14, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Cordeiro, M.H.; Serna, V.A.; Ebbert, K.; Butler, L.M.; Sinha, S.; Mills, A.A.; Woodruff, T.K.; Kurita, T. Rescue of Platinum-Damaged Oocytes from Programmed Cell Death through Inactivation of the P53 Family Signaling Network. Cell Death Differ. 2013, 20, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Livera, G.; Petre-Lazar, B.; Guerquin, M.-J.; Trautmann, E.; Coffigny, H.; Habert, R. p63 Null Mutation Protects Mouse Oocytes from Radio-Induced Apoptosis. Reproduction 2008, 135, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, R.; Tsuchihara, K.; Wilhelm, M.; Fujitani, M.; Rufini, A.; Cheung, C.C.; Khan, F.; Itie-Youten, A.; Wakeham, A.; Tsao, M.; et al. TAp73 Knockout Shows Genomic Instability with Infertility and Tumor Suppressor Functions. Gene Dev. 2008, 22, 2677–2691. [Google Scholar] [CrossRef]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-Deficient Mice Have Neurological, Pheromonal and Inflammatory Defects but Lack Spontaneous Tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Nair, D.M.; Romero, M.; Serna, V.A.; Koleske, A.J.; Woodruff, T.K.; Kurita, T. Transient Inhibition of p53 Homologs Protects Ovarian Function from Two Distinct Apoptotic Pathways Triggered by Anticancer Therapies. Cell Death Differ. 2019, 26, 502–515. [Google Scholar] [CrossRef]
- Elson, A.; Wang, Y.; Daugherty, C.J.; Morton, C.C.; Zhou, F.; Campos-Torres, J.; Leder, P. Pleiotropic Defects in Ataxia-Telangiectasia Protein-Deficient Mice. Proc. Natl. Acad. Sci. USA 1996, 93, 13084–13089. [Google Scholar] [CrossRef]
- Liu, Q.; Guntuku, S.; Cui, X.-S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 Is an Essential Kinase That Is Regulated by Atr and Required for the G2/M DNA Damage Checkpoint. Gene Dev. 2000, 14, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Bernassola, F.; Ranalli, M.; Yee, K.; Zong, W.X.; Corazzari, M.; Knight, R.A.; Green, D.R.; Thompson, C.; Vousden, K.H. p73 Induces Apoptosis via PUMA Transactivation and Bax Mitochondrial Translocation. J. Biol. Chem. 2004, 279, 8076–8083. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Type of Knockout | Phenotypes | References | |
|---|---|---|---|---|
| Bcl-2 | Global KO | Pro-survival BCL-2 family member | Decrease in the number of primordial follicles in ovaries at PND 42 | [8] |
| Global transgenic overexpression | Increase in the number of primordial follicles at PND 8 but not retained in adulthood | [9] | ||
| Oocyte-specific overexpression | Decrease in follicular atresia at PND 42 but no significant phenotypic differences in neonatal ovaries between PNDs 1 and 7 | [10,11] | ||
| Bcl-X (Bcl2l1) | Reduction in Bcl-X (both isoforms: Bcl-XL and Bcl-Xs) gene expression | Pro-survival BCL-2 family member | Drastic decrease in primordial follicle numbers at PND 19 but no significant phenotypic differences in ovaries in later study using conditional knockout | [12,13] |
| Mcl-1 | Oocyte-specific KO | Pro-survival BCL-2 family member | Decrease in primordial oocyte reserve at PND 7 | [14] |
| Decrease in the numbers of primordial follicles, primary follicles and secondary follicles at 3 months which can be rescued by co-deletion of Bax | ||||
| Bcl-w (Bcl2l2, Kiaa0271) | Global KO | Pro-survival BCL-2 family member | Fertile and no abnormalities reported in the ovary | [15] |
| Diva (Bcl2l10, Bcl-b, Boo) | Global KO | Pro-survival BCL-2 family member | Fertile and no abnormalities reported in the ovary | [16] |
| A-1 (Bcl2a1a, Bfl-1) | Global KO | Pro-survival BCL-2 family member | Fertile and no abnormalities reported in the ovary | [17] |
| Bad (Bbc6) | Global KO | Pro-apoptotic BH3-only protein | Ovarian phenotypes not reported | [18] |
| Bik (Biklk, Blk, Nbk) | Global KO | Pro-apoptotic BH3-only protein | Fertile and no abnormalities reported in the ovary | [19] |
| Bid | Global KO | Pro-apoptotic BH3-only protein | Ovarian phenotypes not reported | [20,21] |
| Puma (Bbc3) | Global KO | Pro-apoptotic BH3-only protein | No ovarian phenotype under non-stressed conditions | [22] |
| Primordial follicle oocytes are resistant to irradiation-induced apoptosis at PND 5 | ||||
| Noxa (Pmaip1) | Global KO | Pro-apoptotic BH3-only protein | No significant differences in ovarian morphology | [22] |
| Puma/Noxa | Global DKO | Pro-apoptotic BH3-only proteins | Puma/Noxa DKO oocytes are more resistant to DNA damage-induced death than Puma single KO oocytes | [22] |
| Protected oocyte death in Dmc1 and Msh5-nulls (oocytes with defective recombination repair) | [23] | |||
| Bim (Bcl2l11) | Global KO | Pro-apoptotic BH3-only protein | Fertile and no abnormalities reported in the ovary | [24] |
| Bim/Bad | Global DKO | Pro-apoptotic BH3-only proteins | Fertile and no abnormalities reported in the ovary | [25] |
| Bim/Bik | Global DKO | Pro-apoptotic BH3-only proteins | Fertile and no abnormalities reported in the ovary | [24] |
| Bim/Bid | Global DKO | Pro-apoptotic BH3-only proteins | Ovarian phenotypes not reported | [26] |
| Bid/Bim/Puma | Global TKO | Pro-apoptotic BH3-only proteins | Ovarian phenotypes not reported | [26] |
| Bmf | Global KO | Pro-apoptotic BH3-only protein | Increase in follicles at PNDs 100, 200, 300, and 400 | [27] |
| Fertile and no abnormalities reported in the ovary | [28] | |||
| Hrk (Bid3, Dp5) | Global KO | Pro-apoptotic BH3-only protein | Fertile and no abnormalities reported in the ovary | [29] |
| Bak (Bak1) | Global KO | Pro-apoptotic BCL2 family member | Fertile and no abnormalities reported in the ovary | [30,31] |
| Bax | Global KO | Pro-apoptotic BCL2 family member | Three times as many primordial follicles at PND 42 and reduced follicular atresia (granulosa cell death) induced by apoptosis. No increase observed in primordial follicles in neonatal ovaries | [32] |
| Global KO | Fertile and no abnormalities reported in the ovary | [31] | ||
| Global KO | Protected oocytes from irradiation-induced death and the lack of Dmc1 and Msh5-nulls (oocytes with defective recombination repair) | [23] | ||
| Bok (Mtd) | Global KO | Pro-apoptotic BCL2 family member | Fertile and no abnormalities reported in the ovary | [31,33] |
| Bok/Bak | Global DKO | Pro-apoptotic BCL2 family members | Fertile and no abnormalities reported in the ovary | [31] |
| Bok/Bax | Global DKO | Pro-apoptotic BCL2 family members | Aged (1-year-old) Bok/Bax DKO females had excess follicles at almost all developmental stages, exacerbating the phenotype caused by Bax single KO | [31] |
| Caspase-3 (Casp3, Cpp32) | Global KO | Protease | No significant differences in ovarian morphology | [34] |
| Caspase-7 (Casp7, Lice2, Mch3) | Global KO | Protease | No significant differences in ovarian morphology | [35] |
| Caspase-2 (Casp2, Ich1, Nedd2) | Global KO | Protease | Increase in the number of primordial follicles at PND 4 | [36] |
| Resistance to apoptosis induced by doxorubicin in young adult mice | ||||
| Caspase-11 (Casp4, Casp11, Caspl, Ich3) | Global KO | Protease | Severely diminished primordial follicle pool at PND 4 which can be rescued by Caspase-2 KO | [37] |
| Caspase-9 (Casp9, Mch6) | Global KO | Protease | At 19.5 d.p.c., when the majority of oocytes complete homologous recombination, the total number of germ cells was noticeably larger in Caspase-9 KO embryos. | [38] |
| Fas (Apt1, Tnfrsf6) | Global KO | Cell death receptor | Increase in secondary follicles | [39] |
| Increase in germ cells/oocytes in prenatal and PND 2 to 14 ovaries | [40] | |||
| Tnfa (Tnf, Tnfsf2) | Global KO | Cell death ligand | Increase in the number of total follicles from PND 4 to 90 | [41] |
| Tnfr1 (Tnfrsf1a) | Global KO | Cell death receptor | No significant differences in ovarian morphology | [42] |
| Tnfr2 (Tnfrsf1b) | Global KO | Cell death receptor | Increase in the number of primary follicles at PND 7 and primordial, primary and preantral follicles at PND 80 | [42] |
| Genes | Type of Mouse Models | Protein Functions | Oocyte Phenotypes | References |
|---|---|---|---|---|
| Chk1 (Chek1) | Conditional aneuploid mutant with 3 copies of Chk1 | Kinase | Increase in primordial, primary and antral follicles in 1.5 months old mice as well as primordial and antral follicles in aged mice | [69] |
| Chk2 (Chek2) | Oocyte-specific KO | Kinase | No difference in 1.5 month old mice and increase in primordial, primary, secondary and antral follicles in aged mice | [69] |
| Global KO | Rescued the infertility of Trip13Gt/Gt (DNA repair deficient) and irradiated females | [68] | ||
| Atm | Global KO | Kinase | Infertile and decrease in oocyte reserve in adult females | [70,71] |
| Atr (Kiaa4069) | Global KO | Kinase | Embryonic lethality due to DNA fragmentation between blastocyst stage and 7.5 d.p.c | [72] |
| Trp53 (Tp53, P53) | Global KO | Transcription factor | Partially rescued the Trip13Gt/Gt (DNA repair deficient) oocytes | [68] |
| Trp63 (Tp63, P63) | Oocyte-specific KO | Transcription factor | Rescued from cisplatin-induced cell death of primordial follicles in ovary (PND 5) after 4 days of culture | [73] |
| Global KO | Rescued from irradiation-induced cell death of primordial follicles in ovary (18.5 d.p.c) | [74] | ||
| TAp63 | Global KO | Transcription factor | Primordial follicles protected from irradiation induced apoptosis at PND 5 in TAp63 KO | [66] |
| TAp73 | Global KO | Transcription factor | Decrease in oocyte reserve and infertility | [75] |
| Trp73 | Global KO | Transcription factor | Infertile but no abnormality in oocytes | [76] |
| Oocyte-specific KO | Transcription factor | Partially rescued from cisplatin-induced cell death of primordial follicles in ovary (PND 5), but not from X-ray induced apoptosis | [77] | |
| Mdm2 | Oocyte-specific KO | Ubiquitin E3 Ligase | Infertile and decrease in healthy secondary and tertiary follicles and increase in atretic primary, secondary follicle population at 5–6 weeks of age. Fertility was restored in Mdm2/p53 DKO | [57] |
| Huwe1 (Kiaa0312, Ureb1) | Oocyte-specific KO | Ubiquitin E3 Ligase | Infertile and less follicles present at 4 weeks of age. Fertility was not restored in Huwe1/p53 DKO | [59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, S.; Kurokawa, M. Regulation of Oocyte Apoptosis: A View from Gene Knockout Mice. Int. J. Mol. Sci. 2023, 24, 1345. https://doi.org/10.3390/ijms24021345
Kaur S, Kurokawa M. Regulation of Oocyte Apoptosis: A View from Gene Knockout Mice. International Journal of Molecular Sciences. 2023; 24(2):1345. https://doi.org/10.3390/ijms24021345
Chicago/Turabian StyleKaur, Sandeep, and Manabu Kurokawa. 2023. "Regulation of Oocyte Apoptosis: A View from Gene Knockout Mice" International Journal of Molecular Sciences 24, no. 2: 1345. https://doi.org/10.3390/ijms24021345
APA StyleKaur, S., & Kurokawa, M. (2023). Regulation of Oocyte Apoptosis: A View from Gene Knockout Mice. International Journal of Molecular Sciences, 24(2), 1345. https://doi.org/10.3390/ijms24021345