
TRPML1-Induced Lysosomal Ca2+ Signals Activate AQP2 Translocation and Water Flux in Renal Collecting Duct Cells


 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
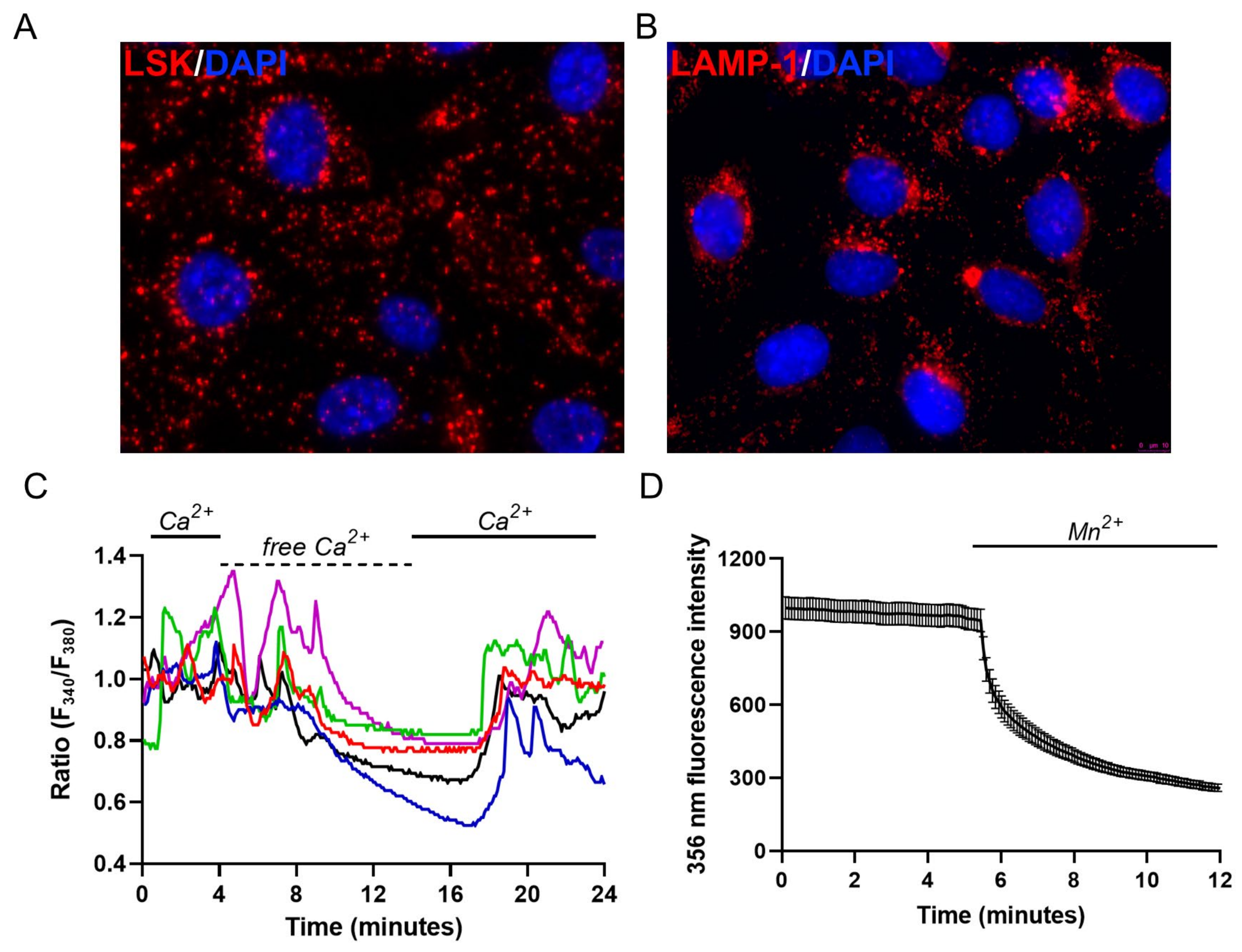
2.1. Endo-Lysosomal System Distribution and Ca2+ Homeostasis Characterization of M1 Cells
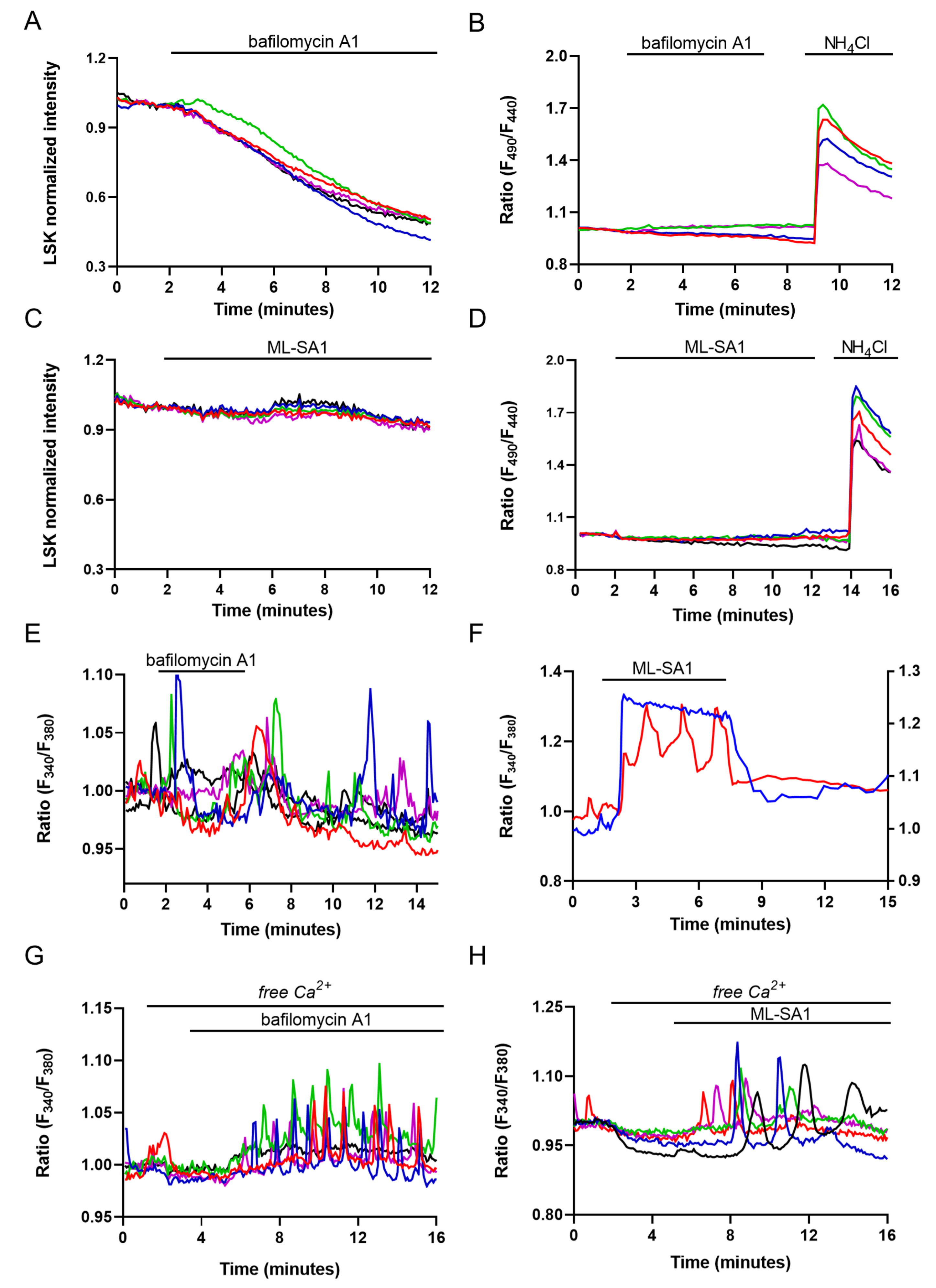
2.2. Evaluation of Lysosomal Ca2+ Signaling Events in M1 Cells
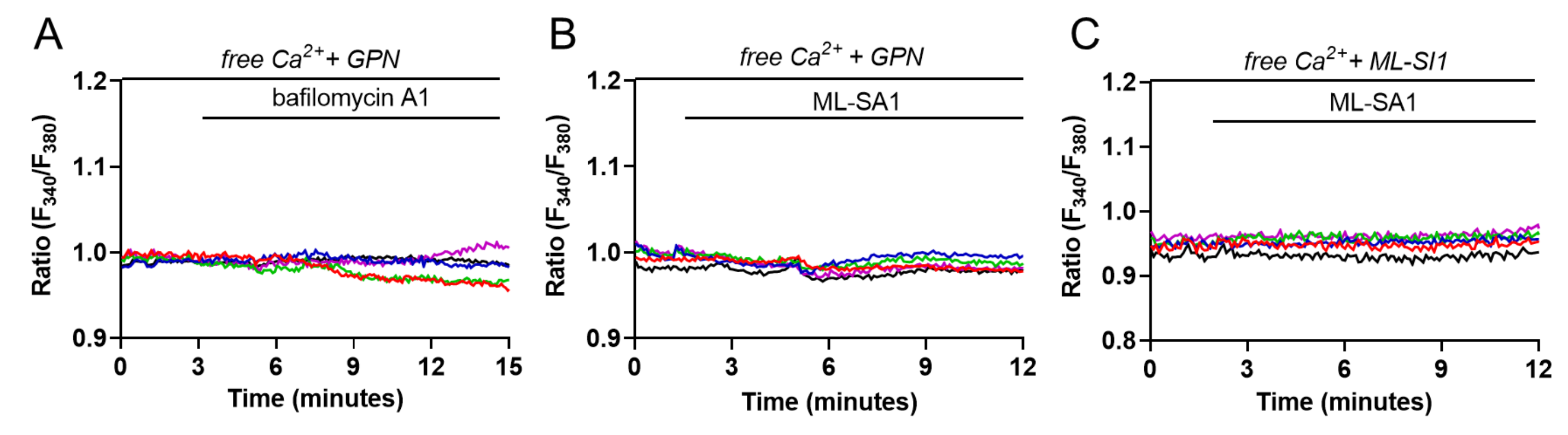
2.3. Evaluation of Lysosome and ER Ca2+ Interplay
2.4. Effect of ML-SA1 and Bafilomycin A1 on AQP2 Intracellular Localization and Cyto-Skeleton Remodeling in MCD4 Cells
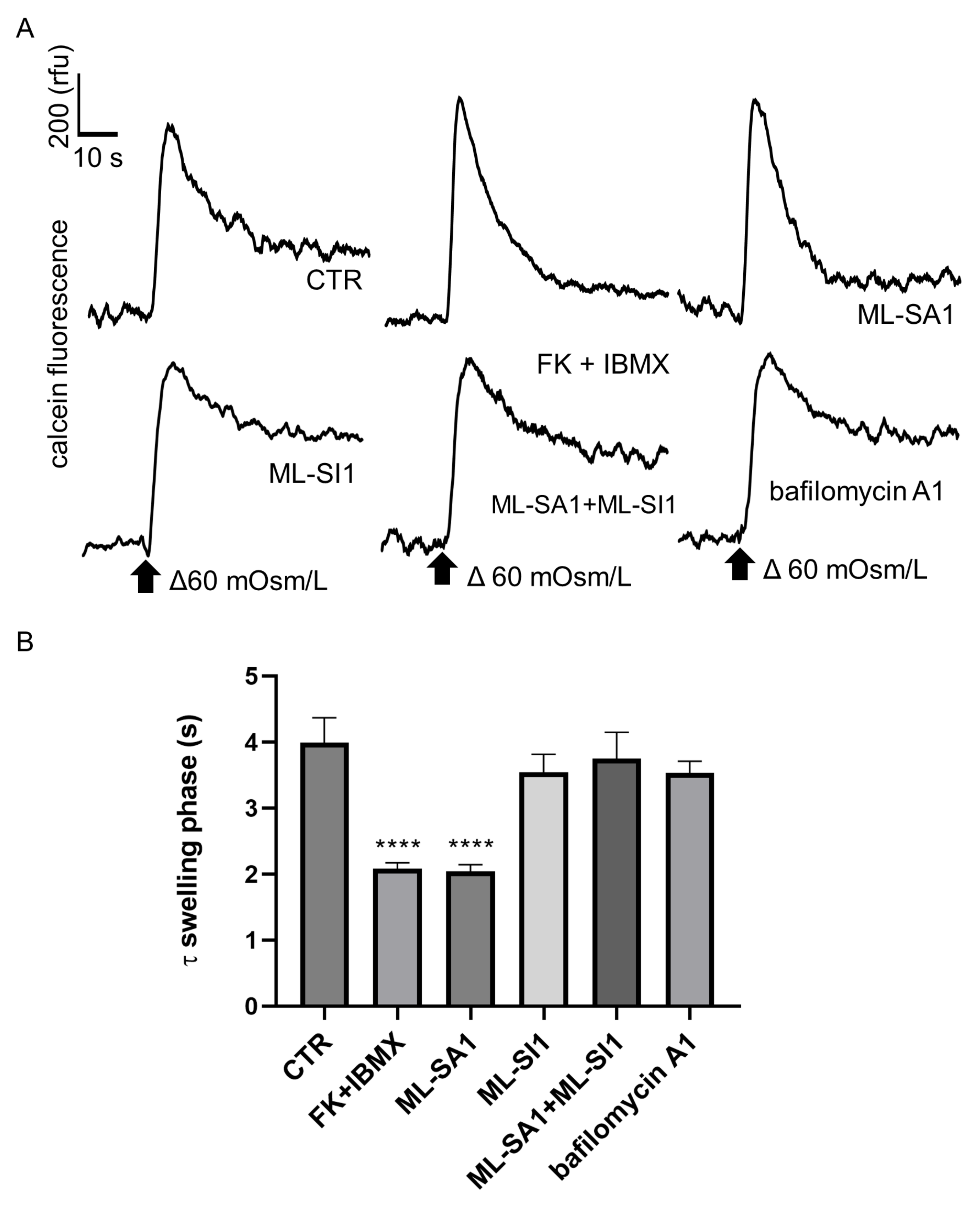
2.5. Effect of ML-SA1 and Bafilomycin A1 on the Swelling Phase of MCD4 Cells under Hypotonic Condition
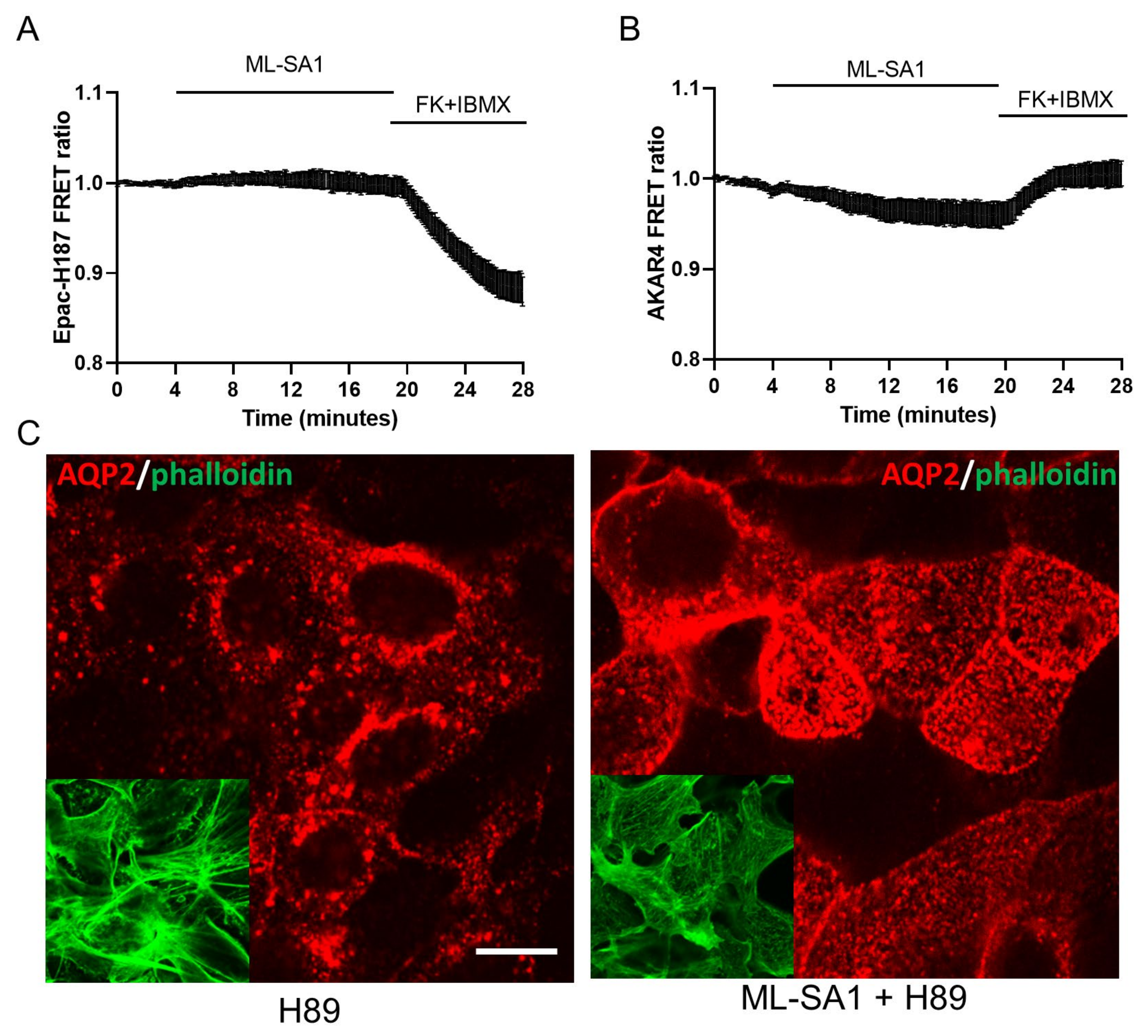
2.6. Effect of TRPML1 Activation on cAMP/PKA and Ca2+/Calcineurin Pathways
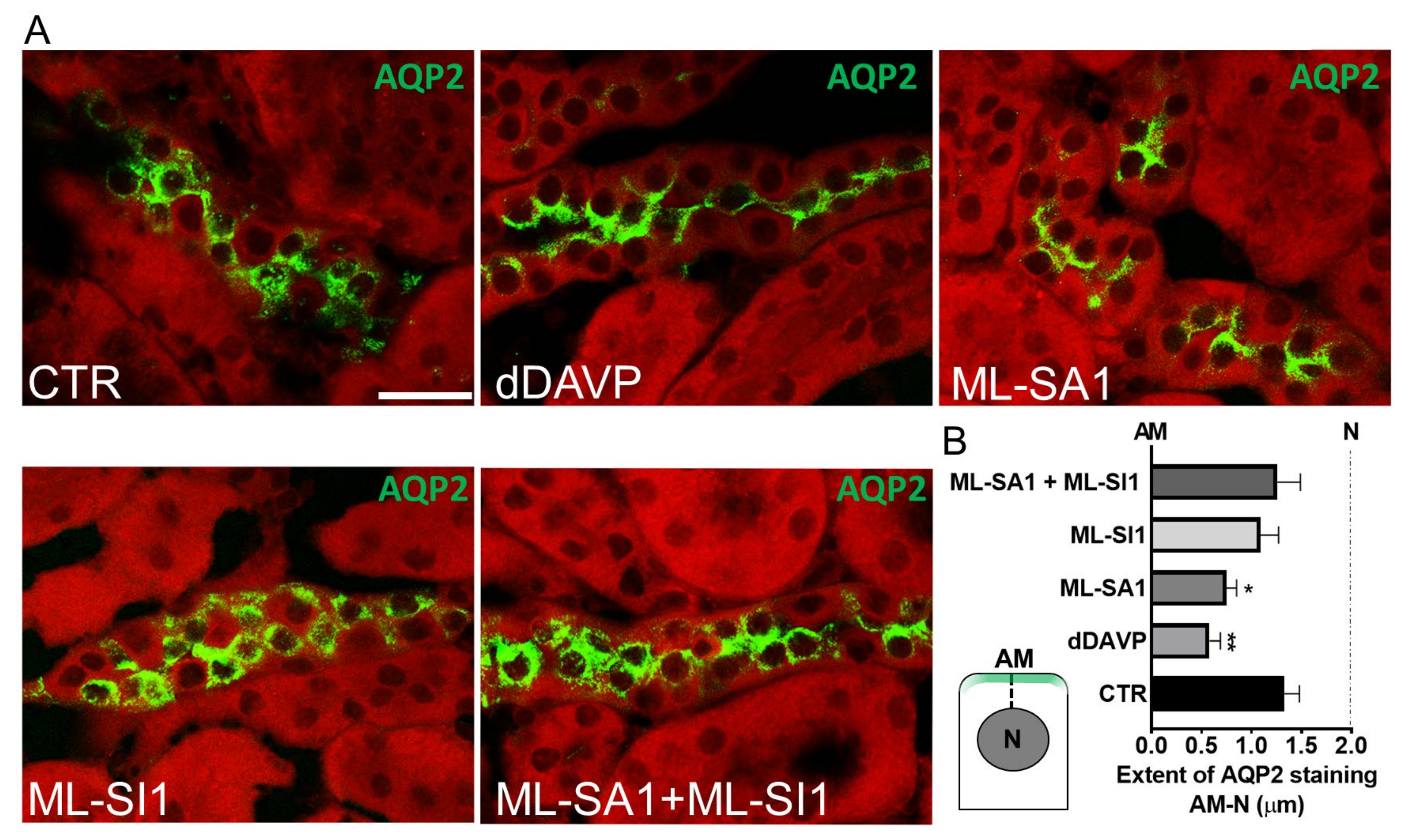
2.7. TRPML1 Stimulation Promotes AQP2 Accumulation on the Apical Membrane of Collecting Duct Cells of Freshly Isolated Kidney Slices
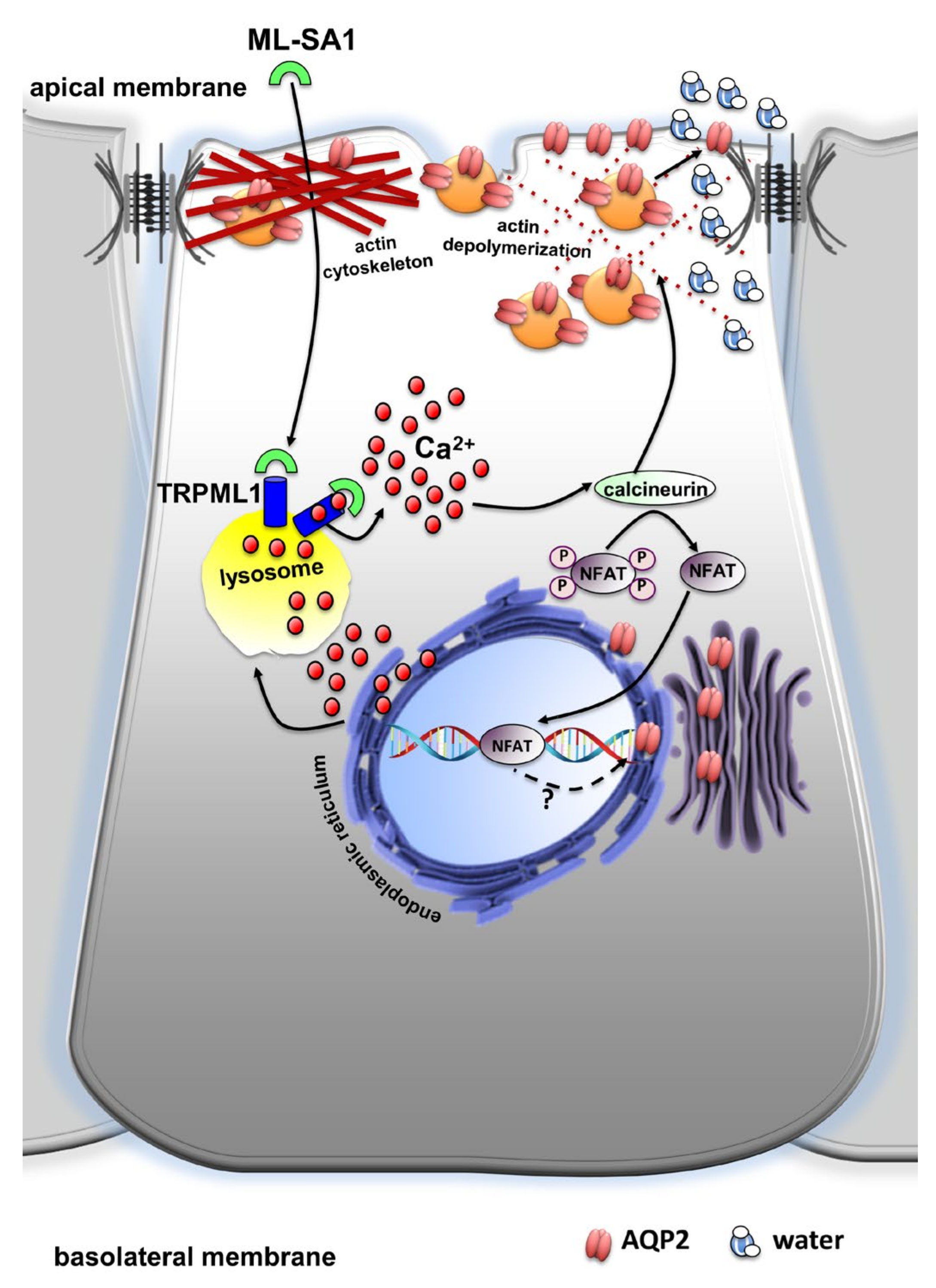
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Evaluation of Cytosolic Ca2+ Levels, Intracellular pH and Lysosomes Visualization
4.4. FRET-Based Measurements of cAMP/PKA in Single M1 Cells
4.5. Evaluation of NFAT-GFP Translocation
4.6. Immunofluorescence and Confocal Microscopy
4.7. Fluorescence-Quenching Assays
4.8. Kidney Slices
4.9. Western Blotting
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saito, A.; Sato, H.; Iino, N.; Takeda, T. Molecular mechanisms of receptor-mediated endocytosis in the renal proximal tubular epithelium. J. Biomed. Biotechnol. 2010, 2010, 403272. [Google Scholar] [CrossRef]
- Milano, S.; Carmosino, M.; Gerbino, A.; Svelto, M.; Procino, G. Hereditary nephrogenic diabetes insipidus: Pathophysiology and possible treatment. An update. Int. J. Mol. Sci. 2017, 18, 2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortenoeven, M.L.A.; Olesen, E.T.B.; Fenton, R.A. Renal Aquaporins in Health and Disease; Springer Nature: Cham, Switzerland, 2020; pp. 1187–1244. [Google Scholar] [CrossRef]
- Surendran, K.; Vitiello, S.P.; Pearce, D.A. Lysosome dysfunction in the pathogenesis of kidney diseases. Pediatr. Nephrol. 2014, 29, 2253–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikrem, Ø.; Skrunes, R.; Tøndel, C.; Leh, S.; Houge, G.; Svarstad, E.; Marti, H.P. Pathomechanisms of renal Fabry disease. Cell Tissue Res. 2017, 369, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Waldek, S.; Feriozzi, S. Fabry nephropathy: A review—How can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014, 15, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atmis, B.; Bayazit, A.K.; Cevizli, D.; Kor, D.; Fidan, H.B.; Bisgin, A.; Kilavuz, S.; Unal, I.; Erdogan, K.E.; Melek, E.; et al. More than tubular dysfunction: Cystinosis and kidney outcomes. J. Nephrol. 2022, 35, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Jamalpoor, A.; Othman, A.; Levtchenko, E.N.; Masereeuw, R.; Janssen, M.J. Molecular Mechanisms and Treatment Options of Nephropathic Cystinosis. Trends Mol. Med. 2021, 27, 673–686. [Google Scholar] [CrossRef]
- Schröder, J.; Lüllmann-Rauch, R.; Himmerkus, N.; Pleines, I.; Nieswandt, B.; Orinska, Z.; Koch-Nolte, F.; Schröder, B.; Bleich, M.; Saftig, P. Deficiency of the Tetraspanin CD63 Associated with Kidney Pathology but Normal Lysosomal Function. Mol. Cell Biol. 2009, 29, 1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.Z.; Yang, Y.; Sun, X.; Dong, X.P. Methods for monitoring Ca2+ and ion channels in the lysosome. Cell Calcium 2017, 64, 20–28. [Google Scholar] [CrossRef]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar] [CrossRef]
- Melchionda, M.; Pittman, J.K.; Mayor, R.; Patel, S. Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J. Cell Biol. 2016, 212, 803–813. [Google Scholar] [CrossRef] [Green Version]
- Ronco, V.; Potenza, D.M.; Denti, F.; Vullo, S.; Gagliano, G.; Tognolina, M.; Guerra, G.; Pinton, P.; Genazzani, A.A.; Mapelli, L.; et al. A novel Ca2+-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca2+ signalling. Cell Calcium 2015, 57, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Gerasimenko, J.V.; Tepikin, A.V.; Petersen, O.H.; Gerasimenko, O.V. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr. Biol. 1998, 8, 1335–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193.e7. [Google Scholar] [CrossRef] [Green Version]
- Patel, S. Function and dysfunction of two-pore channels. Sci. Signal. 2015, 8, re7. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.T.Y.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.E.; Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013, 126, 60–66. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; De Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J. Cell Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca2+ Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers 2019, 11, 542. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, B.S.; Eden, E.R.; Hockey, L.N.; Yates, E.; Futter, C.E.; Patel, S. An Endosomal NAADP-Sensitive Two-Pore Ca2+ Channel Regulates ER-Endosome Membrane Contact Sites to Control Growth Factor Signaling. Cell Rep. 2017, 18, 1636. [Google Scholar] [CrossRef] [Green Version]
- Faris, P.; Casali, C.; Negri, S.; Iengo, L.; Biggiogera, M.; Maione, A.S.; Moccia, F. Nicotinic Acid Adenine Dinucleotide Phosphate Induces Intracellular Ca2+ Signalling and Stimulates Proliferation in Human Cardiac Mesenchymal Stromal Cells. Front. Cell Dev. Biol. 2022, 10, 874043. [Google Scholar] [CrossRef]
- Thakore, P.; Pritchard, H.A.T.; Griffin, C.S.; Yamasaki, E.; Drumm, B.T.; Lane, C.; Sanders, K.M.; Feng Earley, Y.; Earley, S. TRPML1 channels initiate Ca2+ sparks in vascular smooth muscle cells. Sci. Signal. 2020, 13, eaba1015. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Yates, E.; Grimm, C.; Schapira, A.H.; Patel, S. Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J. Cell Sci. 2016, 129, 3859–3867. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 2021, 12, 27. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca2+ signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Waller-Evans, H.; Peterneva, K.; Platt, F.M. Endolysosomal calcium regulation and disease. Biochem. Soc. Trans. 2010, 38, 1458–1464. [Google Scholar] [CrossRef] [Green Version]
- Kiselyov, K.; Yamaguchi, S.; Lyons, C.W.; Muallem, S. Aberrant Ca2+ handling in lysosomal storage disorders. Cell Calcium 2010, 47, 103. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, A.J.; Davis, L.C.; Galione, A. Imaging approaches to measuring lysosomal calcium. Methods Cell Biol. 2015, 126, 159–195. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [Green Version]
- Procino, G.; Gerbino, A.; Milano, S.; Nicoletti, M.C.M.C.; Mastrofrancesco, L.; Carmosino, M.; Svelto, M. Rosiglitazone promotes AQP2 plasma membrane expression in renal cells via a Ca-dependent/cAMP-independent mechanism. Cell Physiol. Biochem. 2015, 35, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Gerbino, A.; Schena, G.; Milano, S.; Milella, L.; Franco Barbosa, A.; Armentano, F.; Procino, G.; Svelto, M.; Carmosino, M. Spilanthol from Acmella Oleracea Lowers the Intracellular Levels of cAMP Impairing NKCC2 Phosphorylation and Water Channel AQP2 Membrane Expression in Mouse Kidney. PLoS ONE 2016, 11, e0156021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milano, S.; Gerbino, A.; Schena, G.; Carmosino, M.; Svelto, M.; Procino, G. Human β3-Adrenoreceptor is Resistant to Agonist-Induced Desensitization in Renal Epithelial Cells. Cell Physiol. Biochem. 2018, 48, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.P. Coupling of vasopressin-induced intracellular Ca2+ mobilization and apical exocytosis in perfused rat kidney collecting duct. J. Physiol. 2002, 538, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef] [Green Version]
- Haller, T.; Dietl, P.; Deetjen, P.; Völkl, H. The lysosomal compartment as intracellular calcium store in MDCK cells: A possible involvement in InsP3-mediated Ca2+ release. Cell Calcium 1996, 19, 157–165. [Google Scholar] [CrossRef]
- Jadot, M.; Colmant, C.; Wattiaux-De Coninck, S.; Wattiaux, R. Intralysosomal hydrolysis of glycyl-L-phenylalanine 2-naphthylamide. Biochem. J. 1984, 219, 965. [Google Scholar] [CrossRef]
- Atakpa, P.; Van Marrewijk, L.M.; Apta-Smith, M.; Chakraborty, S.; Taylor, C.W. GPN does not release lysosomal Ca2+ but evokes Ca2+ release from the ER by increasing the cytosolic pH independently of cathepsin C. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Yuan, Y.; Kilpatrick, B.S.; Gerndt, S.; Bracher, F.; Grimm, C.; Schapira, A.H.; Patel, S. The lysosomotrope GPN mobilises Ca2+ from acidic organelles. J. Cell Sci. 2021, 134, jcs256578. [Google Scholar] [CrossRef] [PubMed]
- Sbano, L.; Bonora, M.; Marchi, S.; Baldassari, F.; Medina, D.L.; Ballabio, A.; Giorgi, C.; Pinton, P. TFEB-mediated increase in peripheral lysosomes regulates store-operated calcium entry. Sci. Rep. 2017, 7, 40797. [Google Scholar] [CrossRef] [PubMed]
- Leser, C.; Keller, M.; Gerndt, S.; Urban, N.; Chen, C.C.; Schaefer, M.; Grimm, C.; Bracher, F. Chemical and pharmacological characterization of the TRPML calcium channel blockers ML-SI1 and ML-SI3. Eur. J. Med. Chem. 2021, 210, 112966. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.D.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Churchill, G.C.; Galione, A. NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3- and cADPR-sensitive Ca2+ stores. EMBO J. 2001, 20, 2666–2671. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.L.; Yip, K.P.; Michea, L.; Kador, K.; Ferraris, J.D.; Wade, J.B.; Knepper, M.A. Regulation of Aquaporin-2 Trafficking by Vasopressin in the Renal Collecting Duct: Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J. Biol. Chem. 2000, 275, 36839–36846. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, L.; Sham, J.S.K.; Yip, K.P. Calcium signaling in vasopressin-induced aquaporin-2 trafficking. Pflugers Arch. Eur. J. Physiol. 2008, 456, 747–754. [Google Scholar] [CrossRef]
- Gustafson, C.E.; Katsura, T.; McKee, M.; Bouley, R.; Casanova, J.E.; Brown, D. Recycling of AQP2 occurs through a temperature- and bafilomycin-sensitive trans-Golgi-associated compartment. Am. J. Physiol. Ren. Physiol. 2000, 278, F317–F326. [Google Scholar] [CrossRef] [Green Version]
- Klarenbeek, J.; Goedhart, J.; Van Batenburg, A.; Groenewald, D.; Jalink, K. Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 2015, 10, e0122513. [Google Scholar] [CrossRef]
- Depry, C.; Allen, M.D.; Zhang, J. Visualization of PKA activity in plasma membrane microdomains. Mol. Biosyst. 2011, 7, 52–58. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.; Mapelli, L.; Canonico, P.L.; Moccia, F.; Genazzani, A.A. Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures. Int. J. Mol. Sci. 2018, 19, 2997. [Google Scholar] [CrossRef] [Green Version]
- Tapella, L.; Soda, T.; Mapelli, L.; Bortolotto, V.; Bondi, H.; Ruffinatti, F.A.; Dematteis, G.; Stevano, A.; Dionisi, M.; Ummarino, S.; et al. Deletion of calcineurin from GFAP-expressing astrocytes impairs excitability of cerebellar and hippocampal neurons through astroglial Na+/K+ ATPase. Glia 2020, 68, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Z.; McDill, B.W.; Kovach, P.A.; Ding, L.; Go, W.Y.; Ho, S.N.; Chen, F. Calcineurin-NFATc signaling pathway regulates AQP2 expression in response to calcium signals and osmotic stress. Am. J. Physiol.-Cell Physiol. 2007, 292, 1606–1616. [Google Scholar] [CrossRef] [Green Version]
- Tomilin, V.N.; Mamenko, M.; Zaika, O.; Ren, G.; Marrelli, S.P.; Birnbaumer, L.; Pochynyuk, O. TRPC3 determines osmosensitive [Ca2+]i signaling in the collecting duct and contributes to urinary concentration. PLoS ONE 2019, 14, e0226381. [Google Scholar] [CrossRef] [Green Version]
- Mamenko, M.; Dhande, I.; Tomilin, V.; Zaika, O.; Boukelmoune, N.; Zhu, Y.; Gonzalez-Garay, M.L.; Pochynyuk, O.; Doris, P.A. Defective store-operated calcium entry causes partial nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2016, 27, 2035–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Wang, X.; Lu, Q.; Ren, H.; Zhang, H. CUP-5, the C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes. Autophagy 2011, 7, 1308–1315. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descazeaud, V.; Mestre, E.; Marquet, P.; Essig, M. Calcineurin regulation of cytoskeleton organization: A new paradigm to analyse the effects of calcineurin inhibitors on the kidney. J. Cell Mol. Med. 2012, 16, 218–227. [Google Scholar] [CrossRef]
- Danthuluri, N.R.; Kim, D.; Brock, T.A. Intracellular alkalinization leads to Ca2+ mobilization from agonist-sensitive pools in bovine aortic endothelial cells. J. Biol. Chem. 1990, 265, 19071–19076. [Google Scholar] [CrossRef]
- Tamma, G.; Procino, G.; Strafino, A.; Bononi, E.; Meyer, G.; Paulmichl, M.; Formoso, V.; Svelto, M.; Valenti, G. Hypotonicity Induces Aquaporin-2 Internalization and Cytosol-to-Membrane Translocation of ICln in Renal Cells. Endocrinology 2007, 148, 1118–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoos, B.A.; Náray-Fejes-Tóth, A.; Carretero, O.A.; Ito, S.; Fejes-Tóth, G. Characterization of a mouse cortical collecting duct cell line. Kidney Int. 1991, 39, 1168–1175. [Google Scholar] [CrossRef] [Green Version]
- Procino, G.; Barbieri, C.; Carmosino, M.; Tamma, G.; Milano, S.; De Benedictis, L.; Mola, M.G.; Lazo-Fernandez, Y.; Valenti, G.; Svelto, M. Fluvastatin modulates renal water reabsorption in vivo through increased AQP2 availability at the apical plasma membrane of collecting duct cells. Pflugers Arch. 2011, 462, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Barbieri, C.; Tamma, G.; De Benedictis, L.; Pessin, J.E.; Svelto, M.; Valenti, G. AQP2 exocytosis in the renal collecting duct–involvement of SNARE isoforms and the regulatory role of Munc18b. J. Cell Sci. 2008, 121, 2097–2106. [Google Scholar] [CrossRef] [Green Version]
- Gerbino, A.; Bottillo, I.; Milano, S.; Lipari, M.; De Zio, R.; Morlino, S.; Mola, M.G.M.G.; Procino, G.; Re, F.; Zachara, E.; et al. Functional Characterization of a Novel Truncating Mutation in Lamin A/C Gene in a Family with a Severe Cardiomyopathy with Conduction Defects. Cell Physiol. Biochem. 2017, 44, 1559–1577. [Google Scholar] [CrossRef]
- Carmosino, M.; Gerbino, A.; Schena, G.; Procino, G.; Miglionico, R.; Forleo, C.; Favale, S.; Svelto, M. The expression of Lamin A mutant R321X leads to endoplasmic reticulum stress with aberrant Ca2+ handling. J. Cell Mol. Med. 2016, 20, 2194–2207. [Google Scholar] [CrossRef] [PubMed]
- Carmosino, M.; Gerbino, A.; Hendy, G.N.G.N.; Torretta, S.; Rizzo, F.; Debellis, L.; Procino, G.; Svelto, M. NKCC2 activity is inhibited by the Bartter’s syndrome type 5 gain-of-function CaR-A843E mutant in renal cells. Biol. Cell 2015, 107, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Jan, C.R.; Ho, C.M.; Wu, S.N.; Tseng, C.J. Mechanism of rise and decay of thapsigargin-evoked calcium signals in MDCK cells. Life Sci. 1998, 64, 259–267. [Google Scholar] [CrossRef]
- Colella, M.; Pozzan, T. Cardiac Cell HypertrophyIn Vitro. Ann. N. Y. Acad. Sci. 2008, 1123, 64–68. [Google Scholar] [CrossRef]
- Colella, M.; Grisan, F.; Robert, V.; Turner, J.D.; Thomas, A.P.; Pozzan, T. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: Role of calcineurin/NFAT as Ca2+ signal integrators. Proc. Natl. Acad. Sci. USA 2008, 105, 2859–2864. [Google Scholar] [CrossRef] [Green Version]
- Post, S.R.; Christian Rump, L.; Zambon, A.; Hughes, R.J.; Buda, M.D.; Paul Jacobson, J.; Kao, C.C.; Insel, P.A. ATP activates cAMP production via multiple purinergic receptors in MDCK-D1 epithelial cells. Blockade of an autocrine/paracrine pathway to define receptor preference of an agonist. J. Biol. Chem. 1998, 273, 23093–23097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deen, P.M.T.; Rijss, J.P.L.; Mulders, S.M.; Errington, R.J.; Van Baal, J.; Van Os, C.H. Aquaporin-2 transfection of Madin-Darby canine kidney cells reconstitutes vasopressin-regulated transcellular osmotic water transport. J. Am. Soc. Nephrol. 1997, 8, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Mola, M.G.; Nicchia, G.P.; Svelto, M.; Spray, D.C.; Frigeri, A. Automated cell-based assay for screening of aquaporin inhibitors. Anal. Chem. 2009, 81, 8219–8229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mola, M.G.; Saracino, E.; Formaggio, F.; Amerotti, A.G.; Barile, B.; Posati, T.; Cibelli, A.; Frigeri, A.; Palazzo, C.; Zamboni, R.; et al. Cell Volume Regulation Mechanisms in Differentiated Astrocytes. Cell Physiol. Biochem. 2021, 55, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Milano, S.; Carmosino, M.; Gerbino, A.; Saponara, I.; Lapi, D.; Dal Monte, M.; Bagnoli, P.; Svelto, M.; Procino, G. Activation of the Thiazide-Sensitive Sodium-Chloride Cotransporter by Beta3-Adrenoreceptor in the Distal Convoluted Tubule. Front. Physiol. 2021, 12, 695824. [Google Scholar] [CrossRef]
- Procino, G.; Carmosino, M.; Milano, S.; Monte, M.D.; Schena, G.; Mastrodonato, M.; Gerbino, A.; Bagnoli, P.; Svelto, M. β3 adrenergic receptor in the kidney may be a new player in sympathetic regulation of renal function. Kidney Int. 2016, 90, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmosino, M.; Rizzo, F.; Procino, G.; Basco, D.; Valenti, G.; Forbush, B.; Schaeren-Wiemers, N.; Caplan, M.J.; Svelto, M. MAL/VIP17, a new player in the regulation of NKCC2 in the kidney. Mol. Biol. Cell 2010, 21, 3985–3997. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scorza, S.I.; Milano, S.; Saponara, I.; Certini, M.; De Zio, R.; Mola, M.G.; Procino, G.; Carmosino, M.; Moccia, F.; Svelto, M.; et al. TRPML1-Induced Lysosomal Ca2+ Signals Activate AQP2 Translocation and Water Flux in Renal Collecting Duct Cells. Int. J. Mol. Sci. 2023, 24, 1647. https://doi.org/10.3390/ijms24021647
Scorza SI, Milano S, Saponara I, Certini M, De Zio R, Mola MG, Procino G, Carmosino M, Moccia F, Svelto M, et al. TRPML1-Induced Lysosomal Ca2+ Signals Activate AQP2 Translocation and Water Flux in Renal Collecting Duct Cells. International Journal of Molecular Sciences. 2023; 24(2):1647. https://doi.org/10.3390/ijms24021647
Chicago/Turabian StyleScorza, Simona Ida, Serena Milano, Ilenia Saponara, Maira Certini, Roberta De Zio, Maria Grazia Mola, Giuseppe Procino, Monica Carmosino, Francesco Moccia, Maria Svelto, and et al. 2023. "TRPML1-Induced Lysosomal Ca2+ Signals Activate AQP2 Translocation and Water Flux in Renal Collecting Duct Cells" International Journal of Molecular Sciences 24, no. 2: 1647. https://doi.org/10.3390/ijms24021647
APA StyleScorza, S. I., Milano, S., Saponara, I., Certini, M., De Zio, R., Mola, M. G., Procino, G., Carmosino, M., Moccia, F., Svelto, M., & Gerbino, A. (2023). TRPML1-Induced Lysosomal Ca2+ Signals Activate AQP2 Translocation and Water Flux in Renal Collecting Duct Cells. International Journal of Molecular Sciences, 24(2), 1647. https://doi.org/10.3390/ijms24021647