Abstract
The mammalian DNA methylation landscape is established and maintained by the combined activities of the two key epigenetic modifiers, DNA methyltransferases (DNMT) and Ten-eleven-translocation (TET) enzymes. Once DNMTs produce 5-methylcytosine (5mC), TET proteins fine-tune the DNA methylation status by consecutively oxidizing 5mC to 5-hydroxymethylcytosine (5hmC) and further oxidized derivatives. The 5mC and oxidized methylcytosines are essential for the maintenance of cellular identity and function during differentiation. Cytosine modifications with DNMT and TET enzymes exert pleiotropic effects on various aspects of hematopoiesis, including self-renewal of hematopoietic stem/progenitor cells (HSPCs), lineage determination, differentiation, and function. Under pathological conditions, these enzymes are frequently dysregulated, leading to loss of function. In particular, the loss of DNMT3A and TET2 function is conspicuous in diverse hematological disorders, including myeloid and lymphoid malignancies, and causally related to clonal hematopoiesis and malignant transformation. Here, we update recent advances in understanding how the maintenance of DNA methylation homeostasis by DNMT and TET proteins influences normal hematopoiesis and malignant transformation, highlighting the potential impact of DNMT3A and TET2 dysregulation on clonal dominance and evolution of pre-leukemic stem cells to full-blown malignancies. Clarification of the normal and pathological functions of DNA-modifying epigenetic regulators will be crucial to future innovations in epigenetic therapies for treating hematological disorders.
1. Introduction
Methylation of cytosine residue in a cytosine-guanine (CpG) dinucleotide is an extensively studied epigenetic mechanism that is catalyzed by DNA methyltransferases (DNMTs), yielding 5-methylcytosine (5mC), the fifth base in DNA. Cytosine methylation serves as a conserved epigenetic mark and exerts profound effects on a spectrum of fundamental processes in cells, including DNA–protein interaction, transcription, chromatin architecture and stability, chromosome segregation, and the integrity of the genome [1,2,3]. As a result, CpG methylation has important implications for key biological processes, including long-term monoallelic repressions such as X chromosome inactivation and genomic imprinting, as well as the silencing of endogenous parasitic sequences (i.e., retrotransposons) and tumor-suppressor genes. In general, high levels of 5mC at promoters can be associated with transcriptional silencing, although their correlation at the genome-wide level is low [1,4]. Promoter methylation can repress transcription by facilitating the formation of a transcriptional repressor complex via the recruitment of 5mC-recognizing proteins such as methyl-CpG-binding proteins (MBDs) or by directly blocking the binding of transcription factors (TFs) [5,6,7,8]. In contrast, gene body methylation tends to be positively correlated with gene transcription [9]. Besides normal biology, DNA methylation is dysregulated under pathological conditions, critically impacting a variety of processes, including every stage of cancer development (i.e., initiation, maintenance, and progression). Indeed, the DNA methylation pattern is recurrently perturbed in cancer and is thus considered a classic hallmark of cancer. Cancer genomes generally display two characteristic patterns of aberrant DNA methylation: a focal increase in DNA methylation at gene promoters (associated with transcriptional silencing of key tumor-suppressor or repair genes) and a global reduction in DNA methylation across the genome (associated with activation of parasitic sequences and genomic instability).
5mC has been considered a very stable base since its discovery. Thus, cytosines were initially thought to exist in either methylated or unmethylated states [10]. However, a further layer of complexity to the covalent modification of cytosine has been revealed as we understood the function of the TET family of dioxygenases. TET proteins fine-tune cytosine methylation by oxidizing the methyl group of 5mC to form 5-hydroxymethylcytosine (5hmC), a process termed DNA hydroxymethylation (Figure 1a). They can further oxidize the hydroxyl group of 5hmC to generate 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). Notably, TET-mediated oxidation of 5mC up to these higher oxidation states (5fC and 5caC) provides routes to the activation of replication-independent demethylation (which will be discussed in detail later). Together, the methylome landscape in the mammalian genome is exquisitely regulated by the complex interplay between DNMT and TET activities.
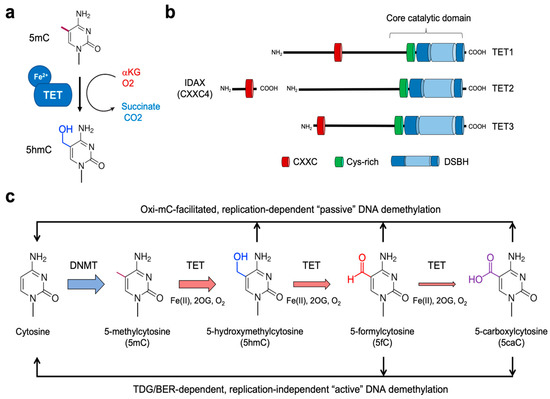
Figure 1.
Role of TET proteins in 5mC oxidation and DNA demethylation. (a) TET proteins belong to the family of Fe2+- and αKG-dependent dioxygenases that oxidize their substrates. All three TET family members successively oxidize 5mC to 5hmC, 5fC, and 5caC. (b) Proteins of the TET family consist of TET1, TET2, and TET3. All three TET proteins have highly conserved catalytic domains at the carboxyl-terminal region, which is composed of the Cys-rich (Cys) and double-stranded β-helix (DSBH) domain. While TET1 and TET3 have the CXXC domain at their amino-terminal regions, TET2 does not contain it. Instead, during evolution, the chromosomal inversion separated the region encoding the CXXC domain of primordial TET2 from that encoding its catalytic domain, giving rise to a unique gene IDAX (which is also called CXXC4). (c) DNMTs methylate cytosine to yield 5mC, which is further oxidized by TET proteins. The oxidized methylcytosines (called oxi-mCs) interfere with DNMT1, thus promoting “passive” DNA demethylation after replication. Moreover, 5fC and 5caC are recognized and cleaved by the DNA repair protein TDG. Then, the resultant abasic sites are repaired by the base-excision repair (BER) pathway, a process called replication-independent “active” DNA demethylation.
Recent large-scale sequencing analyses have successfully revealed a comprehensive catalog of mutational signatures in a wide variety of cancers, thereby facilitating the identification and functional characterization of candidate cancer-causing driver mutations [11,12,13,14]. In hematological neoplasms, numerous somatic mutations recurrently occur in the genes encoding various epigenetic modifiers, including histone/DNA modification enzymes and chromatin remodelers. DNMT3A and TET genes are among the genes most frequently mutated in clonal hematopoiesis and hematologic cancers. Thus, in this review, we briefly review recent progress in our understanding of how both enzymes contribute to the DNA methylation homeostasis, normal hematopoiesis, and malignant transformation, focusing our discussion on the potential molecular mechanisms underlying hematological oncogenesis driven by DNMT3A and TET2 dysregulation.
2. Maintenance of Cytosine Methylation Homeostasis by DNMT and TET Proteins
2.1. Establishing and Maintaining the Mammalian Methylation Landscape
Epigenetic modifications imposed on the mammalian genome confer stability and diversity to the functional state of cells by creating chemically stable but reversible marks that have a direct effect on the local gene activity. DNA cytosine methylation is a central epigenetic modification that is faithfully inherited from parent to daughter cells, a feature that is critical for the preservation of specific gene expression programs and cellular identity across cell divisions [15,16]. On the other hand, DNA methylation is highly mutagenic as 5mC undergoes rapid deamination to thymine, causing C-to-T transition [17]. This inherent mutability of methylated cytosines results in a much lower frequency of the CpG dinucleotides (3~8% of all cytosines) in the genome than expected while increasing a natural source of genetic variations to facilitate the emergence of novel heritable epimutations and epialleles.
Enzymes of the DNMT family are the “writers” of cytosine methylation that catalytically remove a methyl group (−CH3) from S-adenosylmethionine (SAM) and put it at the 5-position (C5) of cytosine to yield 5mC [18,19,20] (Figure 1). Although the mammalian genome shows profound asymmetry in terms of the distribution of CpG-rich and CpG-poor regions, DNMTs typically catalyze symmetrical methylation of cytosine in a 5′-CpG-3′ dinucleotide on both strands of DNA [18,19,20]. DNA methylation patterns are relatively stable in most cell types [21], with over 80% of CpG sites being methylated [22]. However, a small fraction of CpG sites is variably methylated in different tissue lineages and predominantly co-localize with distal cis-regulatory elements (CREs), particularly enhancers and TF binding sites [23].
During early embryogenesis, the de novo DNA methyltransferases DNMT3A and DNMT3B initially deposit the methylation marks on unmethylated templates. Once established, the canonical maintenance methyltransferase DNMT1 ensures the somatic inheritance of the pre-existing methylation patterns via post-replicative methylation of the nascent DNA strand. During the S phase of the cell cycle, DNA replication machinery does not copy 5mC on the parental strand onto the newly synthesized daughter strand, resulting in hemimethylated DNAs. Then, DNMT1 localized to the replication fork restores the symmetrical methylation by methylating the nascent strand. DNMT1 has a marked preference for hemimethylated CpGs due to its physical association with the ubiquitin-like plant homeodomain and RING finger domain-containing protein 1 (UHRF1; also known as NP95) and the proliferating cell nuclear antigen (PCNA) [24,25,26,27,28]. The vast majority of hemimethylated CpG sites are methylated very rapidly within 20 min of replication, although a small fraction of them remain stably hemimethylated and are inherited at CCCTC-binding factor (CTCF)/cohesin-binding sites that regulate chromatin assembly [29]. The failure of maintenance methylation due to impaired expression or function of DNMTs results in replication-dependent “passive” demethylation.
The long-standing view on the divergent functions of DNMT family members as either maintenance (DNMT1) or de novo (DNMT3A/3B) methylases, respectively, has recently been challenged. DNMT1 has been shown to possess a de novo methyltransferase activity in vitro and in vivo [30,31], which is particularly important for the stable repression of retrotransposons [31]. Moreover, DNMT1 alone is not capable of handling maintenance methylation entirely [32,33]. Intriguingly, DNMT3A and DNMT3B exhibit similar activities toward unmethylated and hemimethylated DNA in vitro and can contribute to the maintenance methylation in many cells, including mouse embryonic stem cells (ESCs), neuronal cells, and hematopoietic cells [32,34,35,36,37]. In the absence of DNMT3A/3B, ESCs show high levels of hemimethylated DNA (~30% of CpG sites) in the repetitive elements [38]. These results suggest that the maintenance of mammalian DNA methylome relies on the combined activities of all three DNMTs: the predominant DNA methylase DNMT1 catalyzes the bulk of methylation at the replication forks, particularly on the hemimethylated DNA in dividing cells, and DNMT3A/3B catalyze ongoing methylation of newly replicated CpG sites to complete methylation at specific chromatin regions such as repeat sequences [39].
2.2. Iterative Oxidation of 5mC and DNA Demethylation by TET Proteins
Until recently, 5mC has been considered a terminal cytosine modification form that either remains a stable base or reverts to cytosine through demethylation. Besides the (replication-dependent) passive demethylation, the dynamicity of 5mC abundance is also controlled by the (replication-independent) active demethylation pathway [40,41]. In mammals, the mechanism underpinning passive demethylation is relatively well elucidated. However, it had remained a mystery how 5mC was actively reversed independently of DNA replication until the landmark discovery of the TET enzyme function as a 5mC oxidase [42].
The TET family of dioxygenases, including TET1, TET2, and TET3, directly influence the methylation states by serving as the 5mC “erasers” (Figure 1b) [42,43,44,45]. In 2009, the Rao group discovered the function of TET1 protein based on its homology to base J-binding proteins (JBPs), the thymidine hydroxylases that catalyze the first step in the biosynthesis of an unusual base called base J (β-d-glucosyl-hydroxymethyl-uracil) in kinetoplastid DNAs [42,46,47]. TET proteins belong to the Fe2+- and α-ketoglutarate (αKG, also known as 2-oxoglutarate)-dependent dioxygenase family [48]. Unlike thymidine hydroxylases that oxidize thymine, TET proteins catalyze in situ hydroxylation of 5mC in the 5mCpG dinucleotide to yield 5hmC through an oxidation reaction requiring molecular oxygen, reduced iron (Fe2+), and tricarboxylic-acid-cycle intermediate αKG (Figure 1a). They first transfer a hydroxyl group (−OH) to the methyl group of 5mC to generate 5hmC by transferring one atom of molecular oxygen to the C5-methyl group of 5mC; meanwhile, αKG undergoes oxidative decarboxylation by the other oxygenic atom, releasing CO2 and succinate as byproducts (Figure 1a) [49]. TET proteins carry out two additional oxidation reactions to sequentially oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) [42,44,45]. All TET members enable successive 5mC oxidation due to the conserved catalytic core domains at their carboxyl-terminal regions (Figure 1b). In line with their strong functional link to DNA methylation, TET orthologues are strictly restricted to metazoan organisms that possess cytosine methylation machinery [47,48]. It has been shown that 5fC and 5caC are produced by iterative actions of TET2 protein in a single encounter with 5mC-containing DNA without releasing 5hmC intermediates and that this catalysis is not significantly affected by the modification status of the complementary CpG sites [50].
All three forms of oxidized methylcytosines (oxi-mCs) play a vital role in all known pathways of DNA demethylation in mammals (Figure 1c) [51,52]. First, oxi-mCs in the template strand impair maintenance methylation by interfering with the DNMT1/UHRF1 complex. Thus, the original DNA methylation is lost after cell division [53,54] unless it is maintained by the other methylases DNMT3A/3B. This oxi-mC-facilitated passive demethylation is considered the principal mechanism for the priming of the promoters or enhancers of lineage-specifying genes in dividing cells (Figure 1c) [55,56,57]. Second, replication-independent 5mC removal primarily implicates TET-mediated 5mC oxidation up to 5fC and 5caC, which are removed from the DNA backbone by the DNA repair enzyme thymine DNA glycosylase (TDG) (Figure 1c). TDG typically remove thymine from T:G mismatches and can also hydrolyze the glycosidic linkage between the sugar moiety of DNA and 5fC/5caC that normally pair with G. This cleavage results in abasic sites that are eventually repaired to unmodified cytosine through a base excision repair (BER) pathway [44,58,59,60]. Thus, TET-catalyzed oxi-mCs are pivotal intermediates in active DNA demethylation.
In addition to its role in maintaining enhancer activity to promote cell fate determination, TET-mediated active DNA demethylation was recently shown to generate endogenous DNA damage, particularly single-strand DNA breaks during the BER process [61]. In many cell types, TET-dependent active demethylation seems to play a minor role in replicating cells compared with passive demethylation. Interestingly, TET2-mediated 5mC oxidation was stalled at 5hmC when a conserved residue (Thr1372) in its active site was mutated [62]. As TDG-dependent active demethylation requires oxidation up to 5fC and 5caC, this TET2 variant would be useful to evaluate to what extent TET-mediated active demethylation contributes to certain cellular processes. Furthermore, oxi-mC intermediates have roles as unique epigenetic marks independently of the DNA demethylation pathway, presumably by influencing the chromatin association of methyl CpG-binding proteins or specific oxi-mC-interacting proteins or other epigenetic mechanisms. Thus, it is also useful to probe the biological functions of 5hmC separately from further oxidation products.
Alternatively, AID/APOBEC family enzymes are shown to deaminate 5mC or 5hmC to uracil or 5-hydroxymethyl-uracil, respectively, which are subsequently reverted to cytosine by BER enzymes [63,64]. TDG seems to act as a common mediator in the various demethylation pathways, and its deficiency indeed disrupts normal methylation patterns [65]. However, the active demethylation in the zygotic genome remains unaffected even in the absence of TDG [66], suggesting that there might be unidentified additional strategies by which cells accomplish active demethylation independent of the TET/TDG-dependent pathway. As a potential mechanism, mouse ESCs are shown to possess a 5caC decarboxylase activity [67], although the responsible enzyme remains to be identified.
3. Epigenetic Regulation of Clonal Hematopoiesis by DNMT and TET Proteins
Clonal hematopoiesis of indeterminate potential (CHIP) refers to the aberrant expansion of hematopoietic cell clones without overt abnormalities such as cytopenia, dysplasia, or neoplasia [68,69,70]. CHIP arises from competition over a long period among long-lived HSCs in the bone marrow. Large cohort studies in humans with advanced age have identified ~20 somatic mutations as potential cell-intrinsic contributors to clonal dominance in CHIP. Most of these mutations typically fall within the three functional categories, including epigenetic modifiers (e.g., DNMT3A, TET2, and ASXL1), splicing factors (e.g., SF3B1 and SRSF2), and regulators of DNA damage response (PPM1D and TP53) [69]. In particular, somatic mutations in epigenetic modifiers are remarkably widespread, with ~70% of all CHIP-associated variants being mutations in DNMT3A, TET2, and ASXL1 (Figure 2).
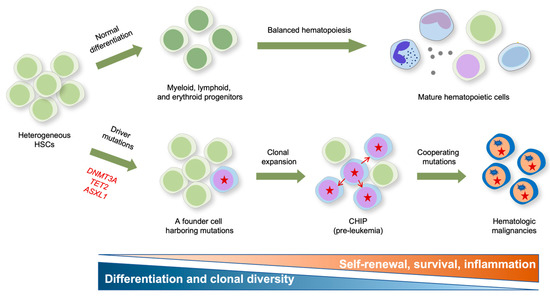
Figure 2.
Impact of epigenetic mutations on the formation and clonal evolution of pre-leukemic stem cells to hematologic malignancies. A model for clonal expansion of hematopoietic stem cells (HSCs) to establish the preleukemic condition that eventually evolves into frank malignancies is shown. Healthy HSCs self-renew and differentiate into multipotent progenitors that give rise to functional tri-lineage hematopoietic cells in the periphery (upper panel). However, aging HSCs acquire somatic mutations in genes encoding the key epigenetic modifiers DNMT3A and TET2 that confer competitive fitness advantage and drive the clonal expansion of mutant HSCs, resulting in CHIP (lower panel). DNMT3A mutation is presumed to mainly influence multipotency (i.e., self-renewal), and TET2 mutations preferentially influence inflammation and myeloid bias, respectively. The pre-leukemic stem cells eventually evolve into full-blown malignancies after acquiring subsequent cooperating mutations in genes encoding FLT3, ASXL1, JAK2, EZH2, NRAS, KIT, RHOA, DNMT3A, SRSF2, AML-ETO1, etc.
Different CHIP mutations are shown to drive clonal expansion with substantially different growth rates, and mutations driving faster clonal growth tend to carry an increased risk of malignancy [71]. While DNMT3A mutant clones preferentially expanded early in life and underwent a slower clonal expansion in old age, TET2 mutations emerged across all ages and induced faster clonal expansion, resulting in TET2 becoming the most prevalent CHIP driver in old age [71]. Consistent with their potential roles in CHIP, DNMT3A and TET2 mutations are early events occurring in HSCs during the clonal evolution to leukemia (Figure 2) [72,73]. Notably, DNMT3A mutations occur more frequently in multipotent HSCs and propagate in all blood lineages, while TET2 mutations occur in more committed progenitors with myeloid potential, suggesting that DNMT3A mutations primarily contribute to multipotency and that TET2 mutations confer a strong myeloid bias [74]. To functionally characterize these variants, many murine models harboring a conditional deletion of these genes in a hematopoietic system have been generated. These animal models show that mutations augmenting HSC self-renewal and fitness, rather than those influencing their balanced differentiation, exert the most potent effect on CHIP in general [75]. In particular, the LOF of DNA methylation regulators DNMT3A and TET2 efficiently drives CHIP by rendering the HSPC more competitive through enhanced self-renewal and restricted differentiation, as described below.
4. DNMTs in Normal and Malignant Hematopoiesis
4.1. DNMTs in HSC Self-Renewal and Lineage Specification
CpG methylation stabilizes the self-renewal and lineage commitment of HSPCs during normal hematopoiesis [76,77,78,79]. Many knockout (KO) studies in mice have demonstrated that constitutive methylation is essential for the maintenance of stemness (i.e., self-renewal and multipotency)-related gene expression as well as the suppression of premature activation of lineage-affiliated genes in HSPCs (Table 1). Despite identical biochemical activities, different DNMT family members exert distinct effects on these processes, although the reason for this is not understood entirely. Loss of DNMT1 in mice, as achieved by conditional gene deletion or the expression of a hypomorphic variant in the DNMT1 KO background, significantly disrupted the homeostasis and self-renewal of HSCs regardless of transplantation stress [76,79]. DNMT1 deficiency led to DNA hypomethylation in HSCs, resulting in widespread transcriptional deregulation. Strikingly, these transcriptional alterations occurred in a lineage-specific manner: myeloerythroid genes (e.g., Gata1, Id2, and Cebpa) were derepressed, whereas lymphoid and stem cell-related genes were downregulated in HSCs, which was supported functionally by markedly skewed differentiation toward myeloerythroid lineages with impaired lymphopoiesis [76]. These observations support the notion that DNA methylation may epigenetically divert lymphoid progenitors from the default differentiation program toward a myeloid lineage.
On the other hand, consistent with the recurrent DNMT3A mutation in CHIP, DNMT3A loss in mice augmented the self-renewal of HSCs, with their differentiation being compromised over serial transplantation on a per-HSC basis (i.e., a lower output of mature blood cells per HSC) [77,80,81]. Compared with DNMT3A loss, DNMT3B LOF displayed similar but milder phenotypes, and simultaneous deletion of DNMT3A and DNMT3B exhibited synergistic effects, causing enhanced HSC self-renewal and a more severe differentiation block [80]. While it remains to be elucidated how DNMT3A loss exerts this profound effect on HSCs, several pieces of evidence suggest that modulation of DNA methylation by DNMT3A at HSC regulatory elements plays a critical role. In DNMT3A-KO HSCs, overall methylation levels remained largely unchanged, and even alterations in methylation were poorly correlated with changes in gene expression in general. However, DNMT3A deficiency seemed to induce notable hypomethylation and derepression of the key multipotency-related genes, including Runx1, Gata3, Pbx1, and Cdkn1a, while downregulating differentiation-associated genes such as Flk2, Ikaros, Sfpi1 (Pu.1), and Mef2c. Intriguingly, DNMT3A loss allowed HSCs to self-renew over at least 12 rounds of transplantation by reducing DNA methylation at enhancers or canyons associated with self-renewal genes [82]. Thus, upon differentiation signals, DNMT3A may methylate and repress a handful of HSC self-renewal genes to allow for downstream differentiation.
4.2. Dysregulation of DNMT3A in Hematologic Malignancies
The DNA methylation abnormalities arising from dysfunctional DNMTs are linked to the initiation and progression of hematological cancers. Unlike DNMT1, somatic mutations in DNMT3A are prevalent in hematologic malignancies of myeloid and lymphoid lineage, including AML (~20%) and myelodysplastic syndromes (MDS; ~10%), and these mutations are associated with a poor prognosis [83]. In AML, DNMT3A mutations are highly enriched for heterozygous point mutations at position R882 (most commonly R882H) within the catalytic domain. The DNMT3AR882 hotspot mutation is a hypomorph that diminishes methyltransferase activity to ~20% of normal levels by disrupting active tetramer formation, thus acting in a dominant negative manner [84]. In addition, multiple nonsense or frameshift mutations are also presumed to produce truncated forms of the DNMT3A enzyme with defective methylase activity [85]. Notably, most CHIP-related DNMT3A mutations are also heterozygous and presumed to be LOF mutations, and these mutations occur all along the length of the gene, although R882 mutations are also frequent [70].

Table 1.
Hematopoietic phenotypes of DNMT-deficient animal models.
Table 1.
Hematopoietic phenotypes of DNMT-deficient animal models.
| Genotype | Major KO Mice Phenotype | Hematologic Malignancy | References |
|---|---|---|---|
| Dnmt1−/chip | Disrupted HSC homeostasis and self-renewal; diminished repopulation capacity; myeloerythroid skewing; derepression of myeloerythroid genes and suppression of lymphoid and stem cell-related genes in HSCs | Not observed | [76] |
| Dnmt1fl/fl Mx1-Cre | Defective HSC self-renewal, BM niche retention, and multilineage differentiation; diminished repopulation capacity; enhanced myeloid lineage gene expression | Not observed | [79] |
| Dnmt3afl/fl Mx1-Cre (competitive transplantation) | Augmented HSC self-renewal and suppressed differentiation over serial transplantation; global hypomethylation and CpG island hypermethylation; increased expression of multipotency genes but reduced expression of differentiation genes in HSCs | Not observed | [77] |
| Dnmt3afl/fl Dnmt3bfl/fl Mx1-Cre (competitive transplantation) | Similar but milder effect in Dnmt3b KO mice; synergistic effects of double KO in enhancing HSC self-renewal; mild global hypomethylation; HSC differentiation block due to activated β-catenin signaling | Not observed | [80] |
| Dnmt3afl/fl Tet2fl/fl Mx1-Cre (competitive transplantation) | Limitless self-renewal of Dnmt3a KO HSC in vivo; exhaustion of Tet2 KO HSC; myeloid skewing and rapid expansion of Tet2 KO progenitors | Not observed | [81] |
| Dnmt3afl/fl Mx1-Cre (competitive transplantation) | Limitless self-renewal of Dnmt3a KO HSC in vivo (>12 rounds of transplantation); focal loss of DNA methylation at self-renewal-associated genes; compromised differentiation potential | Not observed | [82] |
| Dnmt3a+/− | Age-associated myeloid skewing and a competitive transplantation advantage | Myeloid malignancy (37.2% of mice at >20 mo; transplantable); no T cell leukemia | [85] |
| EμSRα-tTA;Teto-Cre; Dnmt3afl/fl | Splenomegaly largely due to expansion of mature B1 B-cells; ~20% decrease in overall gene body methylation; hypomethylation of repetitive elements; CLL and T-cell malignancies in Dnmt3a/b double KO mice | Chronic lymphocytic leukemia (100%, median survival, 371 days, B-cell malignancy); no myeloid malignancy | [86] |
| Dnmt3afl/fl Mx1-Cre (non-competitive transplantation) | Lineage-specific methylation aberrations; acquisition of spontaneous mutations, including Kras; accelerated Nras-driven neoplasia by DNMT3A loss | Myeloid malignancy (MDS (24.39%), AML (19.51%); B-ALL and T-ALL (9.75%); median survival, 321 days | [87] |
| Dnmt3afl/fl Mx1-Cre (non-competitive transplantation) | Bone marrow failure; enhanced HSC serial replating capacity; dysfunctional myeloid and erythroid development; acquisition of c-Kit mutation | MDS-like disease (76%, median survival, 328 days; transplantable); MPD (16%) and AML (8%); cooperation with c-Kit mutation in the development of acute leukemia (median survival, 67 days) | [88] |
| Dnmt3afl/fl Mx1-Cre | Increased HSCPC self-renewal; cytopenia; impaired erythropoiesis; myeloproliferation | MDS/MPN (median survival, 48.6 wk; transplantable) | [89] |
| Tetracycline-inducible Dnmt3b knock-in | Impaired leukemia development and leukemia stem cell function; widespread DNA hypermethylation; | Blockade of Myc-Blc2- or MLL-AF9-induced leukemogenesis | [90] |
| BMT after retroviral overexpression of DNMT3AR882H | Aberrant expression of hematopoiesis-related genes with corresponding changes in gene body methylation | CMML-like disease (100% of mice) | [91] |
Animal studies have shown that the LOF of DNMT3A can drive the transformation from HSPCs to different malignancies (Table 1). DNMT3A loss was not enough to immediately trigger the transformation of murine hematopoietic cells, but its long-term ablation predisposes mice to develop heterogeneous malignancies [85,86,87,88], suggesting that DNMT3A mutations possess moderate leukemogenic potential in vivo. Mice heterozygous for germ-line deletion of the Dnmt3a allele showed myeloid skewing and a competitive transplantation advantage and eventually developed transplantable myeloid malignancies after a long latency [85]. Likewise, conditional deletion of Dnmt3a in the hematopoietic system also resulted in lethal, fully penetrant, and transplantable myeloproliferative neoplasms (MPNs) with a median survival of 48.6 weeks [89]. In a separate study, conditional deletion of Dnmt3a in stem/progenitor cells led to chronic lymphocytic leukemia (CLL) with a median survival of 371 days, which was accelerated by the combined deletion of Dnmt3b [86]. However, no myeloid malignancies were observed in these animals. In contrast, forced expression of Dnmt3b in mice significantly delayed leukemogenesis induced by either Myc-Bcl2 or MLL-AF9 [90].
When Dnmt3a KO HSCs were transplanted into lethally irradiated mice without healthy bone marrow cells, all the mice died within one year of a range of hematologic malignancies such as MDS, AML, and T- and B-cell acute lymphoblastic leukemia (Table 1), the diseases also frequently observed in patients with DNMT3A mutations, and the sick mice acquired a variety of cooperating mutations [87,88]. Furthermore, chimeric mice reconstituted with bone marrow cells overexpressing the DNMT3AR882H mutant also developed myeloproliferation resembling CMML due to impaired gene expression and DNA methylation [88,91]. Most of the DNMT-disrupted murine models display alterations in the DNA methylation patterns and transcriptional programs, but it is unclear whether the altered methylome is directly attributed to the transcriptional changes and malignant transformation. As many genes related to HSC self-renewal or dysregulated in leukemia (e.g., HoxA9, Meis1, and Evi1) were under the control of large undermethylated domains termed “canyons” whose boundaries were eroded in the absence of DNMT3A [37], DNMT3A-mediated methylation of canyon borders may also contribute to the suppression of transformation. Furthermore, it also remains to be clarified how DNMT3A inactivation results in diverse types of malignancies. Given that different types of diseases in the same DNMT3A KO model display distinct lineage-specific methylation profiles [87], deficiency of DNMT3A may induce pre-leukemia, which then transform into different types of leukemia depending on additional hits.
5. TET Proteins in Normal and Malignant Hematopoiesis
5.1. Impaired TET Expression or Function in Myeloid and Lymphoid Malignancies
Although genes encoding TET1 and TET3 are rarely mutated in hematopoietic diseases, TET2 frequently undergoes somatic mutation, affecting both lymphoid and myeloid lineages [92,93,94]. TET2 mutations are also the second most common mutations in CHIP. TET2 mutations are distributed along the length of its coding region, and many missense mutations are relatively clustered in the catalytic domain, mostly resulting in the LOF of the enzyme. TET2 mutations are prevalent in a range of myeloid malignancies, including AML (~23%), MDS (~25%), MPN (~13%), and CMML (~50%), and also in lymphoid malignancies, including T cell lymphoma (~11.9%) and B cell lymphoma (~2%) [92,93,94].
Particularly, TET2 mutations are highly recurrent events in peripheral T-cell lymphoma (PTCL) such as angioimmunoblastic T cell lymphoma (AITL; 33~63%) and PTCL, not otherwise specified (PTCL-NOS; 20~36%) [95,96,97]. Based on the transcriptional profiles, AITL is a highly aggressive form of PTCL driven by malignant cells derived from follicular helper T (Tfh) cells, and TET2 mutations are more common in a subgroup of PTCL-NOS displaying Tfh-like features. Thus, TET2 mutations in PTCL are thought to be associated with Tfh differentiation [96]. In PTCL, particularly AITL, mutations in RHOA, DNMT3A, and IDH2 genes are also common, and TET2 mutations often co-exist with these mutations [97,98,99,100], consistent with a notion that TET2 mutations may cause preleukemic conditions and require additional mutations to drive full-blown diseases.
TET2 mutations are also frequent in B-cell malignancies, particularly in diffuse large B-cell lymphoma (DLBCL; 6~12%), the most common type of non-Hodgkin’s lymphoma arising from germinal center B cells. Notably, in an assay to quantify 5hmC in the genome of patients with various hematologic malignancies, levels of 5hmC in a significant proportion of patients with wild-type (WT) TET2 (and also WT TET1 and TET3) were as low as those from patients with TET2 mutations [43]. This suggests that TET proteins can be inactivated even without mutations in their coding region, presumably through impaired expression or function of TET mRNAs or proteins. The potential mechanisms are extensively reviewed in [92,93,94]. Importantly, future studies are necessary to resolve whether functional inactivation of TET proteins via non-mutational venues also contributes to CHIP.
5.2. Context-Dependent Function of TET1 and TET3
Accumulated evidence indicates that individual TET family members have distinct impacts on HSC self-renewal, lineage specification, and differentiation (Table 2). Dysregulation of specific members results in oncogenesis toward distinct types of malignancies. Despite low expression in hematopoietic tissues and rare mutations in hematologic neoplasms, TET1 is indispensable for normal and malignant hematopoiesis. TET1 can promote or antagonize transformation depending on the context. As observed in many solid cancers [101], TET1 acts as a tumor suppressor in B-cell malignancy [102]. In non-Hodgkin’s B cell lymphomas, such as DLBCL or follicular lymphoma (FL), TET1 was epigenetically silenced through promoter hypermethylation. TET1 loss resulted in DNA hypermethylation in murine HSPCs and disrupted the expression of many genes implicated in B lineage specification, chromosomal maintenance, and DNA repair [102]. As a result, Tet1 KO mice were predisposed to increased self-renewal, DNA damage accumulation, and lymphoid skewing, eventually developing B-cell lymphoma after a long latency. However, it remains to be determined whether TET1 loss induces lymphoid skewing by influencing transcriptional priming in HSCs and also why lymphoid lineage cells are specifically susceptible to TET1 LOF. Given that TET1 mutations are rare in CHIP, even though they seem to increase HSC self-renewal, TET1 LOF occurring independently of mutations may be implicated in driving clonal hematopoiesis.
In contrast, TET1 can also act as an oncogene during leukemogenesis, particularly in T-cell acute lymphoblastic leukemia (T-ALL). TET1 was directly activated by MLL fusion proteins and enhanced oncogenic transcriptional programs involving HOXA9, MEIS1, and PBX3, thus facilitating leukemogenesis [103,104]. Furthermore, TET1 expression was higher in human T-ALL cell lines and clinical samples [105,106] than in normal bone marrow or B-ALL samples. TET1 depletion significantly disrupted the proliferation of human T-ALL cells in vitro and in vivo by impairing the expression of many oncogenes and DNA repair genes [106]. Interestingly, the PARP inhibitor Olaparib substantially reduced TET1 expression and blocked the leukemic growth of T-ALL cells. In addition, increased TET1 expression was associated with the poor survival of patients with cytogenetically normal acute myeloid leukemia (CN-AML) [107], suggesting that TET1 may play a role as an oncogene in AML. It remains to be fully elucidated how TET1 exerts contrasting effects in different types of hematopoietic malignancies.
Tet3 KO mice did not display any significant hematopoietic abnormalities [92], and a recent study showed that aged Tet3 KO mice harboring haploinsufficiency of the Tet2 allele ultimately developed AML after a long latency [108]. Interestingly, in the leukemic mice, the remaining Tet2 allele was lost during the development of AML. These results suggest the tumor suppressor function of TET3 in malignant hematopoiesis. In contrast, TET3 expression was shown to be aberrant in AML patients, with its depletion suppressing the growth of AML cells in vitro and in vivo. The enforced expression of TET3 substantially impaired myeloid, but not erythroid, colony formation, suggesting its oncogenic roles. Further studies are required to precisely define the role of TET3 in oncogenesis.
5.3. TET2 in HSC Self-Renewal and Lineage Commitment
Despite the antagonistic biochemical activities, with DNMT3A yielding the 5mC mark and TET2 erasing it, deletion of Tet2 in mice paradoxically leads to similar phenotypic outcomes as a DNMT3A LOF in terms of enhanced HSC self-renewal, clonal hematopoiesis, impeded differentiation (on a per-HSC basis), and oncogenesis [43,81,94,109] (Table 2). Although there are subtle differences in the degree of their impacts when compared in parallel, the overall direction of the phenotypic changes is the same in both murine models. Transplantation of Tet2 KO bone marrow cells or Dnmt3a KO HSCs showed augmented peripheral blood chimerism in a cell-intrinsic manner [77,95,110,111,112]. The loss of TET2 and DNMT3A influenced HSC self-renewal to a different extent: in serial transplantation assays, the ability of TET2 KO HSCs to self-renew was transiently increased during early passages of transplantation but decreased up to the level in WT HSCs after the third transplantation, although DNMT3A KO HSCs could regenerate almost indefinitely in vivo [81,82]. Furthermore, while DNMT3A loss more specifically affected HSCs [77,80,81,82], TET2 deficiency exhibited a broader effect on HSPCs. Indeed, the primary impact of the TET2 LOF seems to be driving skewed myeloid differentiation of committed progenitors rather than long-term HSCs [81], in agreement with frequent occurrences of TET2 mutations in myeloid-primed progenitors in CHIP [74].
In contrast, a recent single-cell RNA-sequencing analysis highlights that the opposing effects of DNMT3A and TET2 loss on the DNA methylation status indeed have antagonistic effects on the early HSPC lineage specification [113]. TET2 loss in HSCs favored differentiation skews toward myelomonocytic over erythroid progenitors, while DNMT3A loss caused an opposite shift. Mechanistically, this disturbed hematopoietic lineage commitment was attributed to opposing biases in transcriptional priming, with TET2 and DNMT3A LOF favoring the myelomonocytic and erythroid lineages, respectively, in uncommitted HSCs. Consistent with the notion that direct inhibition of TF binding is considered the primary mode of gene silencing by DNA methylation [114,115], the chromatin accessibility of key lineage-determining TFs was particularly susceptible to methylation changes, and strikingly, its sensitivity was determined by the CpG density of the binding motifs [113]. As the TF binding motif had a higher CpG enrichment, it was more readily inactivated by hypermethylation. Interestingly, the DNA-binding motifs of erythroid TFs had a higher CpG content than those of myelomonocytic TFs. Thus, erythroid TFs (e.g., TAL1 and KLF1) were strongly inactivated by TET2 loss-induced hypermethylation, with an opposite effect being observed in DNMT3A loss-induced hypomethylation. However, myelomonocytic TFs (e.g., IRF8 and SP1) were not significantly affected due to their low CpG content in their binding sites. As a result, TET2 loss caused myelomonocytic skews in HSC priming, whereas DNMT3A loss induced erythroid skews. Thus, DNMT3A and TET2 exert antagonistic effects on genome-wide methylation in HSCs, which is connected to differentiation skews through the differences in CpG enrichment of the TF binding site.
Table 2.
Hematopoietic phenotypes of TET-deficient animal models.
Table 2.
Hematopoietic phenotypes of TET-deficient animal models.
| Genotype | Major KO Mice Phenotype | Hematologic Malignancy | References |
|---|---|---|---|
| Tet1−/− | Increased HSC self-renewal; skewed differentiation toward B lineage; enhanced colony formation in vitro; accumulation of DNA damage | B-cell lymphoma (median survival, 22 mo) | [102] |
| Tet1−/−; bone marrow transfer after retroviral expression of shTet1 | TET1 induction by MLL fusions | Delayed MLL-AF9-induced leukemogenesis | [103] |
| Tet3fl/fl Vav-Cre | Normal tri-lineage differentiation; augmented repopulation capacity | Not observed | [92] |
| Tet2fl/+ Mx-Cre; Tet3fl/+ Mx-Cre; Tet2fl/fl Tet3fl/fl Mx-Cre | Inactivation of nontargeted Tet2 or Tet3 allele in AMLs in the single KO mice | AML in Tet2/3 double KO (median survival, ~10.7 wk); AML with a longer latencies in Tet2 or Tet3 single KO (median survival, ~27 wk) | [108] |
| Tet2fl/fl Mx-Cre or Vav-Cre | Limited HSC self-renewal in serial transplantation; profound myeloid skewing | Myeloid malignancy (MPD); accelerated Flt3ITD-driven AML development | [81] |
| Tet2 gene trap | Enhanced self-renewal and long-term repopulating capacity of fetal liver HSCs; myeloid skewing | Not observed | [109] |
| Tet2 gene trap; Tet2fl/fl Mx1-Cre | Expansion of HSPC and myeloid progenitors; competitive repopulation advantage; myeloid expansion | CMML-like disease (gene trap) | [95] |
| Tet2−/− | Expansion of HSPC and myeloid progenitors; competitive repopulation advantage; skewed differentiation toward myeloid lineage in vitro | Not observed | [110] |
| Tet2 gene trap | Expansion of HSPC and myeloid progenitors; competitive repopulation advantage; profound leukocytosis | Myeloid malignancy (~30% of KO mice; CMML, MPN, MDS, etc.) | [111] |
| Tet2fl/fl Vav-Cre | Expansion of HSPC and myeloid progenitors; competitive repopulation advantage | CMML-like disease | [112] |
| Tet2 gene trap (transplantation of fetal liver cells) | Anemia, lymphopenia, thrombocytopenia, dysplasia of myeloid cells | MDS- or CMML-like diseases | [116] |
| Tet2fl/fl Vav1-Cre or LysM-Cre | Suppression of leukemogenesis by WT, but not catalytically inactive TET2 mutant | CMML (50%) or MPD (33.3%) in Tet2fl/fl Vav1-Cre; no malignancy in Tet2fl/fl LysM-Cre | [117] |
| Tet2fl/fl Tet3fl/fl Mx1-Cre or CreERT2 | Rapid myeloid expansion; strong myeloid skewing; fully-penetrant, transplantable, lethal myeloid leukemia within 3–7 wk | Myeloid leukemia (100%, transplantable, median survival, 1 mo) | [118] |
| Tet2−/−, Tet2mut/mut | Distinct gene expression profiles in both models | Myeloid (44.4%) and lymphoid (38.9%) diseases in Tet2−/− mice; myeloid malignancy (78.5%) in Tet2mutmut mice | [119] |
| Tet2 gene trap | Outgrowth of Tfh-like cells in the spleen; lymphomas with similar gene expression patterns as Tfh cells; aberrant DNA methylation and hydroxymethylation | T-cell lymphoma with Tfh features (median survival, ~67 wk) | [120] |
| Tet2fl/fl Vav-Cre or CD19-Cre | Defective class switch recombination and affinity maturation; germinal center hyperplasia; impaired plasma cell differentiation; mimics CREBBP mutant | Not observed | [121] |
| Tet2fl/fl Vav-Cre | Hypermethylation in germinal center B cells; impaired B-cell TF by loss of enhancer 5hmC | Not observed | [122] |
| Tet2fl/fl CD19-Cre | B-cell accumulation; abnormalities in the B1-cell subset; acquisition of AID-mediated mutations in Tet2 KO tumors | B-cell malignancy (50% of mice) | [123] |
| Tet1−/− Tet2−/− | Increased common lymphoid progenitor and B-cell colony formation; increased short-term, but not long-term, repopulation capacity | B-cell malignancy (median survival, 20 mo, transplantable) | [124] |
| Tet2fl/fl Tet3fl/fl CD19-Cre | Increased G-quadruplexes and R-loops; increased DNA double-strand breaks at immunoglobulin switch regions | B-cell lymphoma (median survival, 20 wk; DLBLC-like); | [125] |
| Tet2fl/fl Tet3fl/fl CreERT2 | Impaired class switch recombination via impaired AID expression; impaired 5hmC modification and chromatin accessibility of super-enhancers in the Aicda locus | Not observed | [56] |
5.4. Dysregulation of TET2 in Hematologic Malignancies
Although TET2 mutations are prevalent in hematologic neoplasms, TET2 mutation alone is insufficient to potently drive hematopoietic transformation (Table 2). Indeed, only a subset of TET2 KO mice developed myeloid and/or lymphoid malignancies with partial penetrance and very long latencies (~2 years) [92,93,94]. Myeloproliferation and lethal neoplasia resembling human CMML, MPN, AML, and MDS were most prominent in mice when Tet2 was deleted in all hematopoietic cells, including HSCs [95,108,111,112,116]. However, with the deletion of Tet2 in differentiated myeloid cells (using LysM-Cre), no malignancies were observed, indicating that the TET2 LOF needs to occur in early HSPCs to initiate hematologic diseases [117]. Myeloid leukemogenesis was strikingly potentiated in mice doubly deficient for TET2 and TET3, resulting in highly aggressive, fully-penetrant, and transplantable myeloid leukemias within three to seven weeks [118]. Furthermore, the catalytic activity of TET2 was initially shown to be essential for the suppression of leukemogenesis [117], but a later study showed that both TET2 KO and catalytic mutant mice developed malignancies with distinct disease spectra: while TET2 KO mice developed both myeloid and lymphoid malignancies, the catalytic mutant mice almost exclusively developed myeloid malignancies [119]. Interestingly, the Tet3 allele was lost during leukemic progression to AML in the TET2 catalytic mutant mice, suggesting that the TET2 catalytic activity might be important for genome stability.
TET2 deficiency also drives lymphomagenesis (Table 2). TET2 depletion in gene trap mice led to T-cell lymphomas with Tfh features after a long latency (median ~67 weeks) [120]. Consistent with frequent TET2 mutations in DLBCL, Tet2 deletion in HSPCs or B cells in mice (using Vav-Cre or CD19-Cre) caused germinal center hyperplasia and impaired plasma cell differentiation by impairing germinal center B cell epigenome and transcriptome [121,122], ultimately developing B-cell lymphoma [123]. Furthermore, mice with combined deletion of Tet1 and Tet2 in HSPCs (using Mx-Cre) developed lethal B cell malignancies and died within 20 months [124]. Furthermore, mice with a combined deletion of Tet2 and Tet3 in B cells (using CD19-Cre) rapidly developed DLBCL-like tumors from germinal center B cells with complete penetrance and a median survival of ~20 weeks [125]. Notably, the expanded cells in these mice robustly accumulated DNA damage associated with increased G-quadruplex and R-loop structures [125].
Intriguingly, preleukemic hematopoietic cells from Csf3r/RUNX1 mutant mice progressed to AML by acquiring CXXC4ITD (ITD, internal tandem duplication) mutation as a second hit [126]. The CXXC4 (also called IDAX) gene was originally part of an ancestral TET2 gene. During evolution, it underwent chromosomal rearrangement and was separated from the TET2 gene, forming a separate gene that encodes the CXXC domain of the ancestral TET2 protein (Figure 1a) [127]. The accumulated CXXC4ITD mutations elevated levels of IDAX proteins by increasing their stability. Consistent with the antagonistic effect of IDAX on TET2 protein levels, as previously reported [127], CXXC4ITD mutations decreased TET2 protein levels, which seemed to drive the malignant transformation to AML [126].
5.5. Cooperation with Additional Mutations
As described previously, TET2 mutations seem to increase the pool of pre-leukemic HSPCs that are susceptible to subsequent mutations (i.e., second hits) to develop into full-blown diseases (Figure 2). Indeed, TET2-mutated cancers often harbor various cooperating mutations in genes encoding FLT3, ASXL1, JAK2, EZH2, NRAS, KIT, RHOA, DNMT3A, SRSF2, AML-ETO1, etc. [92,93,128,129]. The outcome of this cooperation has been functionally evaluated in TET2 KO mice harboring one of these mutations, such as Flt3ITD [130], Asxl1 [131], JAK2V617F [132], Ezh2 [133], Nras [134], KITD816V [135], RhoAG17V [136], DNMT3AR882H [137], SRSF2P95H [138], AML-ETO [139], etc. Overall, when combined with a TET2 LOF, these mutations substantially accelerated the development of various types of hematologic neoplasms with significantly shortened latencies; major phenotypes of the mice are summarized in [93].
It is notable that despite the opposing effects of DNMT3A and TET2 loss in shaping early myeloid versus lymphoid biases of early progenitors, loss of either protein in mice leads to similar long-term outcomes in terms of development of lethal malignancies [81,108]. However, TET2- and DNMT3A KO mice responded differently to even the same cooperating mutations: when combined with Flt3ITD mutation, TET2 KO mice died more rapidly of mostly MPNs, while DNMT3A KO mice survived longer but eventually developed mixed phenotype acute leukemia [81]. Despite the epistatic relationship in the DNA methylation-hydroxymethylation pathway, DNMT3A and TET2 mutations are often concurrent in lymphoma and leukemia patients [97,98,99,100], suggesting that both enzymes sometimes work in parallel to produce a common result. Consistent with this, the combined deletion of Dnmt3a and Tet2 in mice displayed synergism to enhance the competitive advantage and expression of lineage-specific TFs in HSCs, eventually resulting in an accelerated progression of multiple malignancies, including CMML-like diseases and B/T-cell lymphoma [140]. A similar synergistic impact was also observed in TET2 KO mice expressing DNMT3AR882H mutation [137]. However, it remains poorly understood why the LOF of DNMT3A and TET2 results in divergent effects in early HSPC commitment but more convergent effects later.
6. TET Modulation of Inflammation in Clonal Hematopoiesis
Chronic low-grade inflammation is a hallmark of aging and thus has gained considerable attention from hematologists due to its potential role as a cell-extrinsic contributor to CHIP (Figure 2). Bone marrow niches play an active role in the initiation and progression of hematologic cancers, particularly myeloid malignancies [141]. Niche-driven pro-inflammatory signals such as tumor necrosis factor α, interleukin-6 (IL-6), and IL-1 create a hostile environment for normal HSCs and can contribute to malignancy by conferring a competitive advantage on HSCs harboring specific mutations [141]. Previous studies have identified inflammation as a key determinant for the selective advantage of TET2 KO HSPC and disease progression. Notably, TET2 KO HSPC resisted inflammatory signals. Upon pro-inflammatory stimuli such as lipopolysaccharide or diabetes-induced hyperglycemia, TET2 KO HSPC and mature myeloid cells were amplified, increasing systemic levels of IL-6 [142,143]. Then, IL-6 led to hyperactivation of the Shp2/Stat3/Morrbid pathway in TET2 KO HSPCs. Given that Morrbid is an anti-apoptotic long noncoding RNA that selectively suppresses pro-apoptotic Bim expression, TET2 loss may provide preleukemic HSPCs with a survival advantage to drive clonal expansion in an inflammatory milieu [142,143]. Likewise, TET2 loss in murine and human HSPCs also augmented clonal advantage under an inflammatory environment containing TNF-α [144]. Another study showed that TET2 deficiency led to systemic bacterial dissemination and elevated IL-6 production by disrupting the integrity of the intestinal barrier, which was critical for preleukemic myeloproliferation. Notably, the TET2 loss-induced myeloproliferation could be substantially reversed using antibiotic treatment or under germ-free conditions [145]. In agreement with this observation, the suppression of gut microbiota-dependent inflammation with antibiotics or pharmacological inhibition of TNF-α also suppressed the expansion of myeloid and lymphoid malignancies in vivo [146]. Together, these results indicate that inflammatory signals confer on TET2-mutant HSPC a competitive advantage to drive clonal expansion. Intriguingly, TET2 acted as a cell-intrinsic suppressor of the inflammatory response in myeloid cells by recruiting HDAC2 to suppress IL-6 [147,148]. Thus, CHIP-associated TET2 mutations may establish a positive feedback loop: the TET2-mutant myeloid cells potentiate CHIP by amplifying the inflammatory environment through elevated IL-6 secretion, which then further augments the competitive advantage and amplification of TET2-mutant HSPCs in the bone marrow by exerting a negative effect on the normal counterparts.
7. Perspectives and Conclusions
Recent systematic studies involving the next-generation sequencing of tumor genomes have shown that mutations in genes encoding two key DNA-modifying enzymes, DNMT3A and TET2, are recurrent events in CHIP as well as in a wide range of hematologic malignancies. These mutations occur at an early HSPC stage, and then the expanded premalignant clones acquire additional mutations to further develop into frank malignancies (Figure 2). Thus, the advancement of next-generation sequencing may enable the early detection of hematologic neoplasms. However, only a subset of aged individuals with CHIP develops full-blown malignancy, so factors determining the progression of CHIP clones to malignancy remain to be fully determined.
The mutational profiling in patient samples and analyses of various murine models mimicking LOF mutations in DNMT3A and TET2, alone or in combination, have made significant progress in our understanding of the underlying molecular mechanisms through which these epigenetic regulators influence normal and malignant hematopoiesis. Nonetheless, there are several key areas in which many unresolved questions remain (Figure 3). Foremost among these would be why DNMT3A and TET2 mutations have profound effects on HSC clonal expansion and progression to malignancies, how they display overlapping and nonoverlapping effects, how they interact with a variety of second hits during clonal evolution to full-blown malignancies, how they work together with cell-extrinsic factors to establish clonal dominance, and whether the LOF in the absence of coding region mutations also leads to similar effects on CHIP and oncogenesis. It will also be interesting to define whether the combined LOF of TET proteins, with TET2 and TET1/3 being inactivated genetically and epigenetically, respectively, display more robust driving effects in CHIP and oncogenesis. Fundamentally, both proteins are crucial regulators of gene expression that control DNA methylation status, TF binding, DNA flexibility and integrity, chromatin architecture and stability, histone modifications, and even three-dimensional genome interactions. Any aberrations occurring in these processes may have critical impacts on the establishment of clonal hematopoiesis and subsequent tumor progression. Therefore, further characterization of the epigenetic regulation of clonal hematopoiesis and malignant transformation is warranted to decipher the precise transformation mechanism.
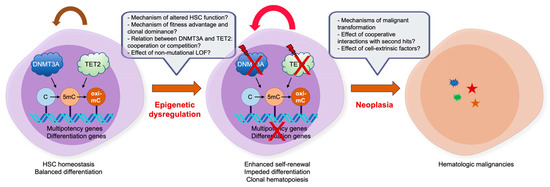
Figure 3.
Impact of DNMT and TET mutations in CHIP and hematologic malignancies. Despite the progress we have made in the last decades to decipher the role of epimutations, such as DNMT3A and TET2 LOF mutations, there are still many outstanding issues. For details, refer to the text.
Author Contributions
Writing—original draft preparation, J.A. and M.K.; writing—review and editing, J.A. and M.K.; supervision, M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by the National Research Foundation grants of Korea (2022R1F1A1066420 to J.A., and 2018R1D1A1B07049676 and 2022R1A2C1011306 to M.K.). M.K. is also supported by the Center for Genomic Integrity, Institute for Basic Science (IBS, IBS-R022-D1).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.; Chen, T. Genetic Studies on Mammalian DNA Methyltransferases. Adv. Exp. Med. Biol. 2022, 1389, 111–136. [Google Scholar] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H.; Bourc’his, D. Transposon silencing and imprint establishment in mammalian germ cells. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 381–387. [Google Scholar] [CrossRef]
- Jansz, N. DNA methylation dynamics at transposable elements in mammals. Essays Biochem. 2019, 63, 677–689. [Google Scholar]
- Xie, M.; Hong, C.; Zhang, B.; Lowdon, R.F.; Xing, X.; Li, D.; Zhou, X.; Lee, H.J.; Maire, C.L.; Ligon, K.L.; et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat. Genet. 2013, 45, 836–841. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Taryma-Lesniak, O.; Sokolowska, K.E.; Wojdacz, T.K. Short history of 5-methylcytosine: From discovery to clinical applications. J. Clin. Pathol. 2021, 74, 692–696. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Programming of DNA methylation patterns. Annu. Rev. Biochem. 2012, 81, 97–117. [Google Scholar] [CrossRef]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Nabel, C.S.; Manning, S.A.; Kohli, R.M. The curious chemical biology of cytosine: Deamination, methylation, and oxidation as modulators of genomic potential. ACS Chem. Biol. 2012, 7, 20–30. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Ooi, S.K.; O’Donnell, A.H.; Bestor, T.H. Mammalian cytosine methylation at a glance. J. Cell Sci. 2009, 122 Pt 16, 2787–2791. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Scholer, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Muller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.S.; Ian, H.I.; Koh, T.W.; Ng, H.H.; Xu, G.; Li, B.F. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 1997, 277, 1996–2000. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef]
- Arita, K.; Ariyoshi, M.; Tochio, H.; Nakamura, Y.; Shirakawa, M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature 2008, 455, 818–821. [Google Scholar] [CrossRef]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Li, Y.; Duan, S.; Bronner, C.; Arrowsmith, C.H.; Dhe-Paganon, S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 2008, 455, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Corces, V.G. Nascent DNA methylome mapping reveals inheritance of hemimethylation at CTCF/cohesin sites. Science 2018, 359, 1166–1170. [Google Scholar] [CrossRef]
- Vertino, P.M.; Yen, R.W.; Gao, J.; Baylin, S.B. De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol. Cell. Biol. 1996, 16, 4555–4565. [Google Scholar] [CrossRef]
- Haggerty, C.; Kretzmer, H.; Riemenschneider, C.; Kumar, A.S.; Mattei, A.L.; Bailly, N.; Gottfreund, J.; Giesselmann, P.; Weigert, R.; Brandl, B.; et al. Dnmt1 has de novo activity targeted to transposable elements. Nat. Struct. Mol. Biol. 2021, 28, 594–603. [Google Scholar] [CrossRef]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Z.; Shi, J.; Zhang, Y.; Yang, S.; Chen, Q.; Song, C.; Geng, S.; Li, Q.; Li, J.; et al. DNA methyltransferases are complementary in maintaining DNA methylation in embryonic stem cells. iScience 2022, 25, 105003. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L. In vivo activity of murine de novo methyltransferases, Dnmt3a and Dnmt3b. Mol. Cell. Biol. 1999, 19, 8211–8218. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef]
- Jeong, M.; Sun, D.; Luo, M.; Huang, Y.; Challen, G.A.; Rodriguez, B.; Zhang, X.; Chavez, L.; Wang, H.; Hannah, R.; et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014, 46, 17–23. [Google Scholar] [CrossRef]
- Liang, G.; Chan, M.F.; Tomigahara, Y.; Tsai, Y.C.; Gonzales, F.A.; Li, E.; Laird, P.W.; Jones, P.A. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell. Biol. 2002, 22, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef]
- Hackett, J.A.; Surani, M.A. DNA methylation dynamics during the mammalian life cycle. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20110328. [Google Scholar] [CrossRef]
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Genest, P.A.; ter Riet, B.; Sweeney, K.; DiPaolo, C.; Kieft, R.; Christodoulou, E.; Perrakis, A.; Simmons, J.M.; Hausinger, R.P.; et al. The protein that binds to DNA base J in trypanosomatids has features of a thymidine hydroxylase. Nucleic Acids Res. 2007, 35, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Abhiman, S.; Aravind, L. Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 2011, 101, 25–104. [Google Scholar]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Song, C.X.; He, C.; Zhang, Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu. Rev. Biochem. 2014, 83, 585–614. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.J.; Liu, M.Y.; Nabel, C.S.; Cao, X.J.; Garcia, B.A.; Kohli, R.M. Tet2 Catalyzes Stepwise 5-Methylcytosine Oxidation by an Iterative and de novo Mechanism. J. Am. Chem. Soc. 2016, 138, 730–733. [Google Scholar] [CrossRef]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Hashimoto, H.; Liu, Y.; Upadhyay, A.K.; Chang, Y.; Howerton, S.B.; Vertino, P.M.; Zhang, X.; Cheng, X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012, 40, 4841–4849. [Google Scholar] [CrossRef] [PubMed]
- Otani, J.; Kimura, H.; Sharif, J.; Endo, T.A.; Mishima, Y.; Kawakami, T.; Koseki, H.; Shirakawa, M.; Suetake, I.; Tajima, S. Cell cycle-dependent turnover of 5-hydroxymethyl cytosine in mouse embryonic stem cells. PLoS ONE 2013, 8, e82961. [Google Scholar] [CrossRef]
- Inoue, A.; Zhang, Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 2011, 334, 194. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.J.; Shukla, V.; Samaniego-Castruita, D.; Gonzalez-Avalos, E.; Chakraborty, A.; Yue, X.; Schatz, D.G.; Ay, F.; Rao, A. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci. Immunol. 2019, 4, eaau7523. [Google Scholar] [CrossRef]
- Lio, C.J.; Yue, X.; Lopez-Moyado, I.F.; Tahiliani, M.; Aravind, L.; Rao, A. TET methylcytosine oxidases: New insights from a decade of research. J. Biosci. 2020, 45, 21. [Google Scholar] [CrossRef] [PubMed]
- Schomacher, L.; Niehrs, C. DNA repair and erasure of 5-methylcytosine in vertebrates. BioEssays 2017, 39, 1600218. [Google Scholar] [CrossRef]
- Maiti, A.; Drohat, A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, X.; Lu, J.; Liang, H.; Dai, Q.; Xu, G.L.; Luo, C.; Jiang, H.; He, C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012, 8, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wu, W.; Callen, E.; Pavani, R.; Zolnerowich, N.; Kodali, S.; Zong, D.; Wong, N.; Noriega, S.; Nathan, W.J.; et al. Active DNA demethylation promotes cell fate specification and the DNA damage response. Science 2022, 378, 983–989. [Google Scholar] [CrossRef]
- Liu, M.Y.; Torabifard, H.; Crawford, D.J.; DeNizio, J.E.; Cao, X.J.; Garcia, B.A.; Cisneros, G.A.; Kohli, R.M. Mutations along a TET2 active site scaffold stall oxidation at 5-hydroxymethylcytosine. Nat. Chem. Biol. 2017, 13, 181–187. [Google Scholar] [CrossRef]
- Morgan, H.D.; Dean, W.; Coker, H.A.; Reik, W.; Petersen-Mahrt, S.K. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: Implications for epigenetic reprogramming. J. Biol. Chem. 2004, 279, 52353–52360. [Google Scholar] [CrossRef]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, X.; Liang, D.; Li, T.; Zhu, P.; Guo, H.; Wu, X.; Wen, L.; Gu, T.P.; Hu, B.; et al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 2014, 15, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Schiesser, S.; Hackner, B.; Pfaffeneder, T.; Muller, M.; Hagemeier, C.; Truss, M.; Carell, T. Mechanism and stem-cell activity of 5-carboxycytosine decarboxylation determined by isotope tracing. Angew. Chem. Int. Ed. 2012, 51, 6516–6520. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.A.; Lopez-Otin, C. Clonal evolution in leukemia. Nat. Med. 2017, 23, 1135–1145. [Google Scholar] [CrossRef]
- Florez, M.A.; Tran, B.T.; Wathan, T.K.; DeGregori, J.; Pietras, E.M.; King, K.Y. Clonal hematopoiesis: Mutation-specific adaptation to environmental change. Cell Stem Cell 2022, 29, 882–904. [Google Scholar] [CrossRef]
- Challen, G.A.; Goodell, M.A. Clonal hematopoiesis: Mechanisms driving dominance of stem cell clones. Blood 2020, 136, 1590–1598. [Google Scholar] [CrossRef]
- Fabre, M.A.; de Almeida, J.G.; Fiorillo, E.; Mitchell, E.; Damaskou, A.; Rak, J.; Orru, V.; Marongiu, M.; Chapman, M.S.; Vijayabaskar, M.S.; et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 2022, 606, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Bourgoin, V.; Mollica, L.; Dube, M.P.; Busque, L. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood 2018, 132, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.; Lin, K.K.; Boles, N.C.; Yang, L.; King, K.Y.; Jeong, M.; Mayle, A.; Goodell, M.A. Less is more: Unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell 2012, 11, 302–317. [Google Scholar] [CrossRef]
- Broske, A.M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 2009, 41, 1207–1215. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, 23–31. [Google Scholar] [CrossRef]
- Tadokoro, Y.; Ema, H.; Okano, M.; Li, E.; Nakauchi, H. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J. Exp. Med. 2007, 204, 715–722. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef]
- Ostrander, E.L.; Kramer, A.C.; Mallaney, C.; Celik, H.; Koh, W.K.; Fairchild, J.; Haussler, E.; Zhang, C.R.C.; Challen, G.A. Divergent Effects of Dnmt3a and Tet2 Mutations on Hematopoietic Progenitor Cell Fitness. Stem Cell Rep. 2020, 14, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.B.; Russler-Germain, D.A.; Ketkar, S.; Verdoni, A.M.; Smith, A.M.; Bangert, C.V.; Helton, N.M.; Guo, M.; Klco, J.M.; O’Laughlin, S.; et al. Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J. Clin. Investig. 2017, 127, 3657–3674. [Google Scholar] [CrossRef]
- Peters, S.L.; Hlady, R.A.; Opavska, J.; Klinkebiel, D.; Pirruccello, S.J.; Talmon, G.A.; Sharp, J.G.; Wu, L.; Jaenisch, R.; Simpson, M.A.; et al. Tumor suppressor functions of Dnmt3a and Dnmt3b in the prevention of malignant mouse lymphopoiesis. Leukemia 2014, 28, 1138–1142. [Google Scholar] [CrossRef]
- Mayle, A.; Yang, L.; Rodriguez, B.; Zhou, T.; Chang, E.; Curry, C.V.; Challen, G.A.; Li, W.; Wheeler, D.; Rebel, V.I.; et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Celik, H.; Mallaney, C.; Kothari, A.; Ostrander, E.L.; Eultgen, E.; Martens, A.; Miller, C.A.; Hundal, J.; Klco, J.M.; Challen, G.A. Enforced differentiation of Dnmt3a-null bone marrow leads to failure with c-Kit mutations driving leukemic transformation. Blood 2015, 125, 619–628. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Lieu, Y.K.; Garrett-Bakelman, F.E.; Spitzer, B.; Glass, J.L.; Shank, K.; Martinez, A.B.; Rivera, S.A.; Durham, B.H.; Rapaport, F.; et al. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia 2016, 30, 1133–1142. [Google Scholar] [CrossRef]
- Schulze, I.; Rohde, C.; Scheller-Wendorff, M.; Baumer, N.; Krause, A.; Herbst, F.; Riemke, P.; Hebestreit, K.; Tschanter, P.; Lin, Q.; et al. Increased DNA methylation of Dnmt3b targets impairs leukemogenesis. Blood 2016, 127, 1575–1586. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.Y.; Dai, Y.J.; Zhang, W.; Zhang, W.N.; Xiong, S.M.; Gu, Z.H.; Wang, K.K.; Zeng, R.; Chen, Z.; et al. DNMT3A Arg882 mutation drives chronic myelomonocytic leukemia through disturbing gene expression/DNA methylation in hematopoietic cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2620–2625. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; An, J.; Pastor, W.A.; Koralov, S.B.; Rajewsky, K.; Rao, A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol. Rev. 2015, 263, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.J.; Yuita, H.; Rao, A. Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood 2019, 134, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; An, J.; Rao, A. DNA methylation and hydroxymethylation in hematologic differentiation and transformation. Curr. Opin. Cell Biol. 2015, 37, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Quivoron, C.; Couronne, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef]
- Lemonnier, F.; Couronne, L.; Parrens, M.; Jais, J.P.; Travert, M.; Lamant, L.; Tournillac, O.; Rousset, T.; Fabiani, B.; Cairns, R.A.; et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 2012, 120, 1466–1469. [Google Scholar] [CrossRef]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.; van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Palomero, T.; Couronne, L.; Khiabanian, H.; Kim, M.Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Couronne, L.; Bastard, C.; Bernard, O.A. TET2 and DNMT3A mutations in human T-cell lymphoma. N. Engl. J. Med. 2012, 366, 95–96. [Google Scholar] [CrossRef]
- Bray, J.K.; Dawlaty, M.M.; Verma, A.; Maitra, A. Roles and Regulations of TET Enzymes in Solid Tumors. Trends Cancer 2021, 7, 635–646. [Google Scholar] [CrossRef]
- Cimmino, L.; Dawlaty, M.M.; Ndiaye-Lobry, D.; Yap, Y.S.; Bakogianni, S.; Yu, Y.; Bhattacharyya, S.; Shaknovich, R.; Geng, H.; Lobry, C.; et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol. 2015, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jiang, X.; Li, Z.; Li, Y.; Song, C.X.; He, C.; Sun, M.; Chen, P.; Gurbuxani, S.; Wang, J.; et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 11994–11999. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jiang, X.; Wang, J.; Li, Y.; Song, C.X.; Chen, P.; Li, S.; Gurbuxani, S.; Arnovitz, S.; Wang, Y.; et al. Identification of MLL-fusion/MYC dash, verticalmiR-26 dash, verticalTET1 signaling circuit in MLL-rearranged leukemia. Cancer Lett. 2016, 372, 157–165. [Google Scholar] [CrossRef]
- Bamezai, S.; Demir, D.; Pulikkottil, A.J.; Ciccarone, F.; Fischbein, E.; Sinha, A.; Borga, C.; Te Kronnie, G.; Meyer, L.H.; Mohr, F.; et al. TET1 promotes growth of T-cell acute lymphoblastic leukemia and can be antagonized via PARP inhibition. Leukemia 2021, 35, 389–403. [Google Scholar] [CrossRef]
- Poole, C.J.; Lodh, A.; Choi, J.H.; van Riggelen, J. MYC deregulates TET1 and TET2 expression to control global DNA (hydroxy)methylation and gene expression to maintain a neoplastic phenotype in T-ALL. Epigenet. Chromatin 2019, 12, 41. [Google Scholar] [CrossRef]
- Wang, J.; Li, F.; Ma, Z.; Yu, M.; Guo, Q.; Huang, J.; Yu, W.; Wang, Y.; Jin, J. High Expression of TET1 Predicts Poor Survival in Cytogenetically Normal Acute Myeloid Leukemia From Two Cohorts. eBioMedicine 2018, 28, 90–96. [Google Scholar] [CrossRef]
- Shrestha, R.; Sakata-Yanagimoto, M.; Maie, K.; Oshima, M.; Ishihara, M.; Suehara, Y.; Fukumoto, K.; Nakajima-Takagi, Y.; Matsui, H.; Kato, T.; et al. Molecular pathogenesis of progression to myeloid leukemia from TET-insufficient status. Blood Adv. 2020, 4, 845–854. [Google Scholar] [CrossRef]
- Kunimoto, H.; Fukuchi, Y.; Sakurai, M.; Sadahira, K.; Ikeda, Y.; Okamoto, S.; Nakajima, H. Tet2 disruption leads to enhanced self-renewal and altered differentiation of fetal liver hematopoietic stem cells. Sci. Rep. 2012, 2, 273. [Google Scholar] [CrossRef]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 14566–14571. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Izzo, F.; Lee, S.C.; Poran, A.; Chaligne, R.; Gaiti, F.; Gross, B.; Murali, R.R.; Deochand, S.D.; Ang, C.; Jones, P.W.; et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet. 2020, 52, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Kaluscha, S.; Domcke, S.; Wirbelauer, C.; Stadler, M.B.; Durdu, S.; Burger, L.; Schubeler, D. Evidence that direct inhibition of transcription factor binding is the prevailing mode of gene and repeat repression by DNA methylation. Nat. Genet. 2022, 54, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef]
- Muto, T.; Sashida, G.; Hasegawa, N.; Nakaseko, C.; Yokote, K.; Shimoda, K.; Iwama, A. Myelodysplastic syndrome with extramedullary erythroid hyperplasia induced by loss of Tet2 in mice. Leuk. Lymphoma 2015, 56, 520–523. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, S.; Zhu, X.; Pan, F.; Li, R.; Zhou, Y.; Yuan, W.; Ni, H.; Yang, F.C.; Xu, M. The catalytic activity of TET2 is essential for its myeloid malignancy-suppressive function in hematopoietic stem/progenitor cells. Leukemia 2016, 30, 1784–1788. [Google Scholar] [CrossRef]
- An, J.; Gonzalez-Avalos, E.; Chawla, A.; Jeong, M.; Lopez-Moyado, I.F.; Li, W.; Goodell, M.A.; Chavez, L.; Ko, M.; Rao, A. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat. Commun. 2015, 6, 10071. [Google Scholar] [CrossRef]
- Ito, K.; Lee, J.; Chrysanthou, S.; Zhao, Y.; Josephs, K.; Sato, H.; Teruya-Feldstein, J.; Zheng, D.; Dawlaty, M.M.; Ito, K. Non-catalytic Roles of Tet2 Are Essential to Regulate Hematopoietic Stem and Progenitor Cell Homeostasis. Cell Rep. 2019, 28, 2480–2490.e4. [Google Scholar] [CrossRef]
- Muto, H.; Sakata-Yanagimoto, M.; Nagae, G.; Shiozawa, Y.; Miyake, Y.; Yoshida, K.; Enami, T.; Kamada, Y.; Kato, T.; Uchida, K.; et al. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J. 2014, 4, e264. [Google Scholar] [CrossRef]
- Dominguez, P.M.; Ghamlouch, H.; Rosikiewicz, W.; Kumar, P.; Beguelin, W.; Fontan, L.; Rivas, M.A.; Pawlikowska, P.; Armand, M.; Mouly, E.; et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov. 2018, 8, 1632–1653. [Google Scholar] [CrossRef] [PubMed]
- Rosikiewicz, W.; Chen, X.; Dominguez, P.M.; Ghamlouch, H.; Aoufouchi, S.; Bernard, O.A.; Melnick, A.; Li, S. TET2 deficiency reprograms the germinal center B cell epigenome and silences genes linked to lymphomagenesis. Sci. Adv. 2020, 6, eaay5872. [Google Scholar] [CrossRef] [PubMed]
- Mouly, E.; Ghamlouch, H.; Della-Valle, V.; Scourzic, L.; Quivoron, C.; Roos-Weil, D.; Pawlikowska, P.; Saada, V.; Diop, M.K.; Lopez, C.K.; et al. B-cell tumor development in Tet2-deficient mice. Blood Adv. 2018, 2, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, L.; Dawlaty, M.M.; Pan, F.; Weeks, O.; Zhou, Y.; Cao, Z.; Shi, H.; Wang, J.; Lin, L.; et al. Combined Loss of Tet1 and Tet2 Promotes B Cell, but Not Myeloid Malignancies, in Mice. Cell Rep. 2015, 13, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Samaniego-Castruita, D.; Dong, Z.; Gonzalez-Avalos, E.; Yan, Q.; Sarma, K.; Rao, A. TET deficiency perturbs mature B cell homeostasis and promotes oncogenesis associated with accumulation of G-quadruplex and R-loop structures. Nat. Immunol. 2022, 23, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Olofsen, P.A.; Fatrai, S.; van Strien, P.M.H.; Obenauer, J.C.; de Looper, H.W.J.; Hoogenboezem, R.M.; Erpelinck-Verschueren, C.A.J.; Vermeulen, M.; Roovers, O.; Haferlach, T.; et al. Malignant Transformation Involving CXXC4 Mutations Identified in a Leukemic Progression Model of Severe Congenital Neutropenia. Cell Rep. Med. 2020, 1, 100074. [Google Scholar] [CrossRef]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Aijo, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lahdesmaki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013, 497, 122–126. [Google Scholar] [CrossRef]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef]
- Tefferi, A. Mutations galore in myeloproliferative neoplasms: Would the real Spartacus please stand up? Leukemia 2011, 25, 1059–1063. [Google Scholar] [CrossRef]
- Shih, A.H.; Jiang, Y.; Meydan, C.; Shank, K.; Pandey, S.; Barreyro, L.; Antony-Debre, I.; Viale, A.; Socci, N.; Sun, Y.; et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 2015, 27, 502–515. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; Lafave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; Shide, K.; Yamaji, T.; Kamiunten, A.; Sekine, M.; Taniguchi, Y.; Hidaka, T.; Kubuki, Y.; Shimoda, H.; Marutsuka, K.; et al. Loss of TET2 has dual roles in murine myeloproliferative neoplasms: Disease sustainer and disease accelerator. Blood 2015, 125, 304–315. [Google Scholar] [CrossRef]
- Muto, T.; Sashida, G.; Oshima, M.; Wendt, G.R.; Mochizuki-Kashio, M.; Nagata, Y.; Sanada, M.; Miyagi, S.; Saraya, A.; Kamio, A.; et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 2013, 210, 2627–2639. [Google Scholar] [CrossRef]
- Kunimoto, H.; Meydan, C.; Nazir, A.; Whitfield, J.; Shank, K.; Rapaport, F.; Maher, R.; Pronier, E.; Meyer, S.C.; Garrett-Bakelman, F.E.; et al. Cooperative Epigenetic Remodeling by TET2 Loss and NRAS Mutation Drives Myeloid Transformation and MEK Inhibitor Sensitivity. Cancer Cell 2018, 33, 44–59.e8. [Google Scholar] [CrossRef]
- Palam, L.R.; Mali, R.S.; Ramdas, B.; Srivatsan, S.N.; Visconte, V.; Tiu, R.V.; Vanhaesebroeck, B.; Roers, A.; Gerbaulet, A.; Xu, M.; et al. Loss of epigenetic regulator TET2 and oncogenic KIT regulate myeloid cell transformation via PI3K pathway. JCI Insight 2018, 3, e94679. [Google Scholar] [CrossRef]
- Zang, S.; Li, J.; Yang, H.; Zeng, H.; Han, W.; Zhang, J.; Lee, M.; Moczygemba, M.; Isgandarova, S.; Yang, Y.; et al. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J. Clin. Investig. 2017, 127, 2998–3012. [Google Scholar] [CrossRef]
- Scourzic, L.; Couronne, L.; Pedersen, M.T.; Della Valle, V.; Diop, M.; Mylonas, E.; Calvo, J.; Mouly, E.; Lopez, C.K.; Martin, N.; et al. DNMT3A(R882H) mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia 2016, 30, 1388–1398. [Google Scholar] [CrossRef]
- Xu, J.J.; Chalk, A.M.; Wall, M.; Langdon, W.Y.; Smeets, M.F.; Walkley, C.R. Srsf2P95H/+ co-operates with loss of TET2 to promote myeloid bias and initiate a chronic myelomonocytic leukemia-like disease in mice. Leukemia 2022, 36, 2883–2893. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015, 29, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018, 23, 833–849.e5. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Lu, X.; Zhang, C.; Nelanuthala, S.; Aguilera, F.; Hadley, A.; Ramdas, B.; Fang, F.; Nephew, K.; Kotzin, J.J.; et al. Hyperglycemia cooperates with Tet2 heterozygosity to induce leukemia driven by proinflammatory cytokine-induced lncRNA Morrbid. J. Clin. Investig. 2021, 131, e140707. [Google Scholar] [CrossRef] [PubMed]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Zeng, H.; He, H.; Guo, L.; Li, J.; Lee, M.; Han, W.; Guzman, A.G.; Zang, S.; Zhou, Y.; Zhang, X.; et al. Antibiotic treatment ameliorates Ten-eleven translocation 2 (TET2) loss-of-function associated hematological malignancies. Cancer Lett. 2019, 467, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70.e13. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).