

Spontaneous Transition of Alkyl Carbocations to Unsaturated Vinyl-Type Carbocations in Organic Solutions


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
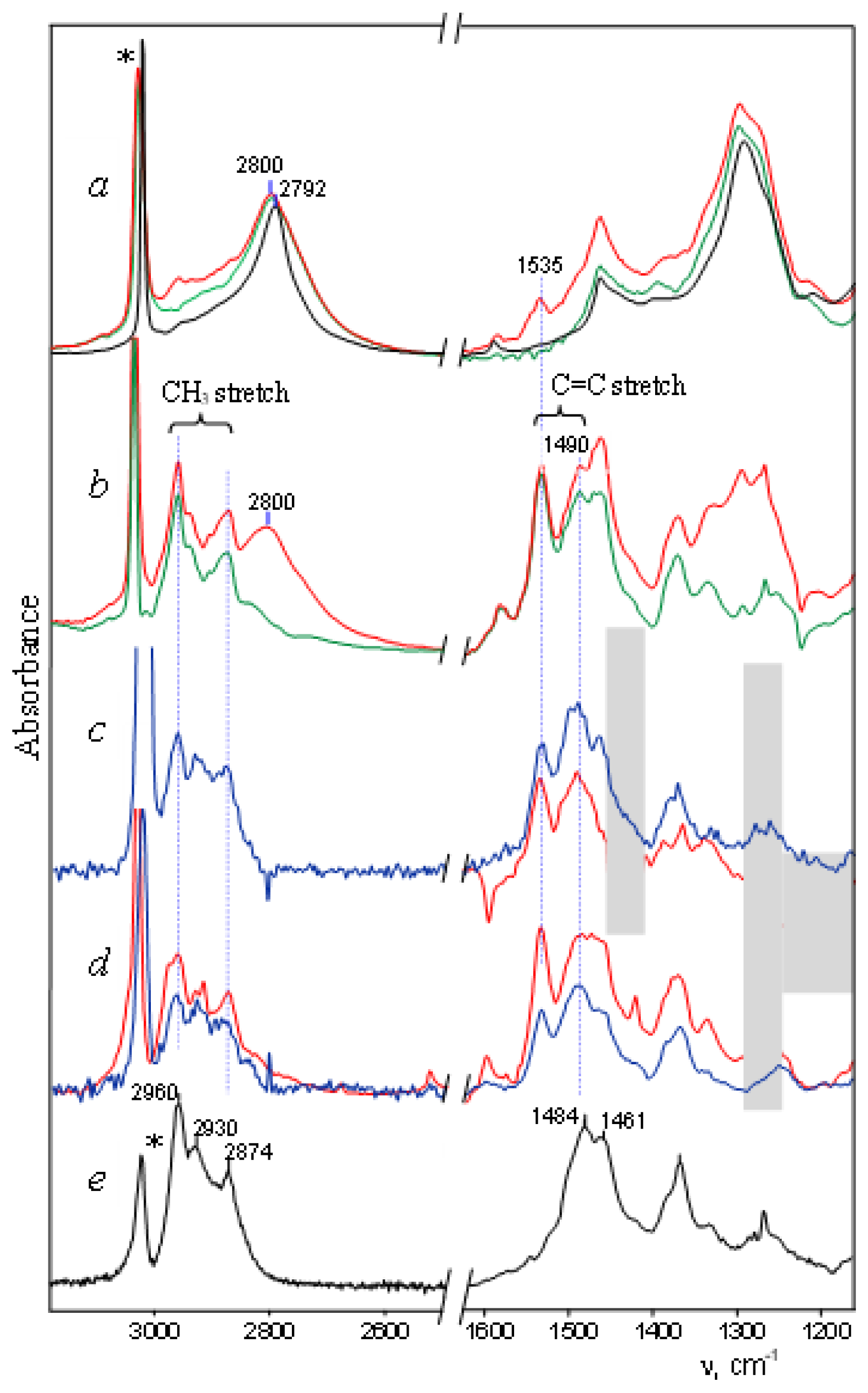
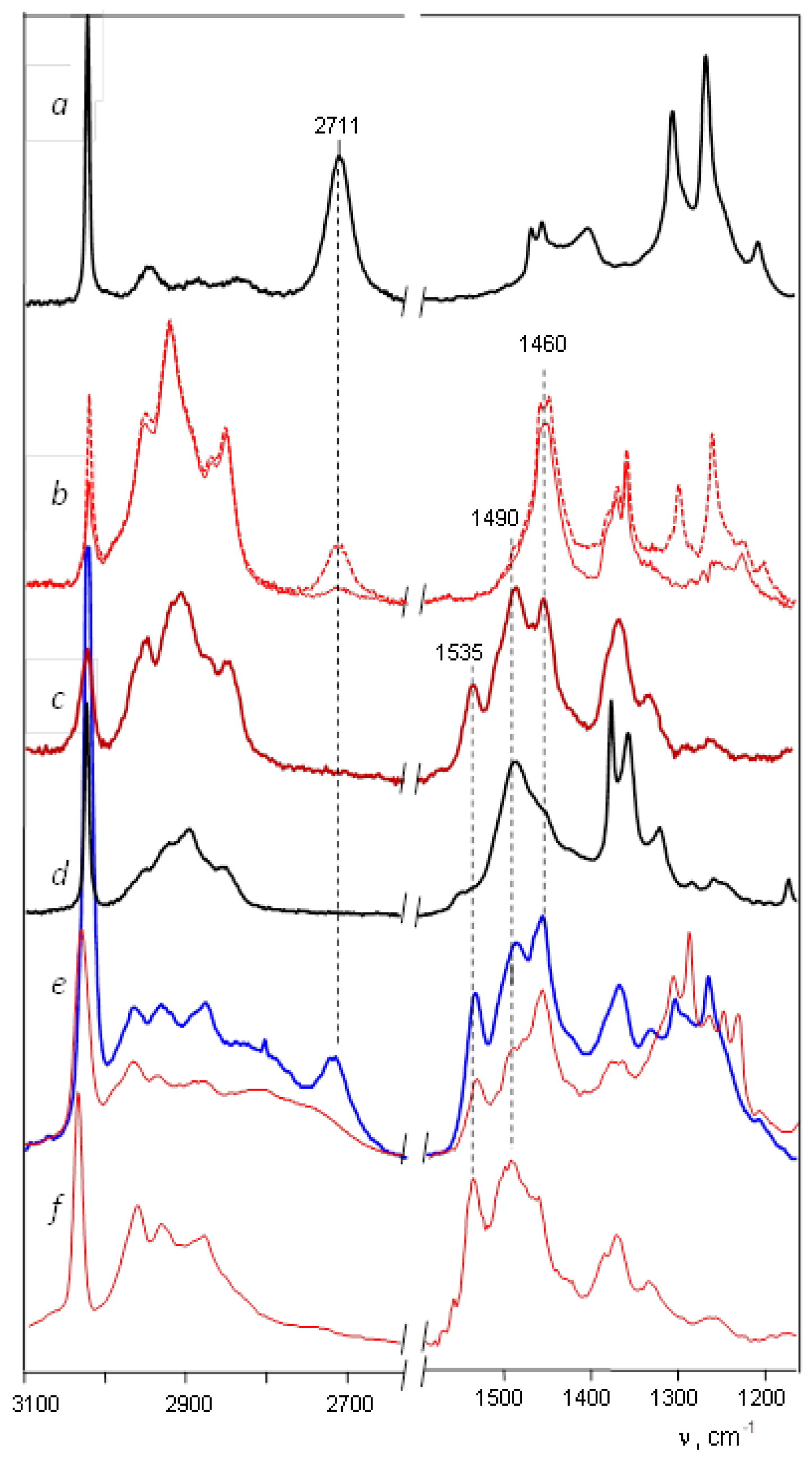
 stretch vibration of allyl cation (CH2CHCH2)+ has a lower frequency [20], at 1303 cm−1. Therefore, it can be assumed that the Y+ cation is of the allyl type. It forms from the alkyl cation of methyl-cyclopentyl+ in a solvent medium and, being unstable, subsequently decomposes into butylene cation Z and other products (Scheme 1). group with close-to-aromatic CC bonds—on which the positive charge is predominantly concentrated—can have a frequency lower than that of the C=C+ stretch; (iii) the CH3 group of the cation, in contrast to methylcyclopentyl+ [23], does not participate in hyperconjugation because it is bonded to the sp2 C atom with a field pz orbital. The Y+ cation is unstable and decomposes yielding two or more unsaturated carbocations. One of them is the butylene cation, the salt of which crystallizes out and allows it to be uniquely identified by X-ray diffraction.
stretch vibration of allyl cation (CH2CHCH2)+ has a lower frequency [20], at 1303 cm−1. Therefore, it can be assumed that the Y+ cation is of the allyl type. It forms from the alkyl cation of methyl-cyclopentyl+ in a solvent medium and, being unstable, subsequently decomposes into butylene cation Z and other products (Scheme 1). group with close-to-aromatic CC bonds—on which the positive charge is predominantly concentrated—can have a frequency lower than that of the C=C+ stretch; (iii) the CH3 group of the cation, in contrast to methylcyclopentyl+ [23], does not participate in hyperconjugation because it is bonded to the sp2 C atom with a field pz orbital. The Y+ cation is unstable and decomposes yielding two or more unsaturated carbocations. One of them is the butylene cation, the salt of which crystallizes out and allows it to be uniquely identified by X-ray diffraction.3. Methods and Materials
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prakash, G.K.S.; Schleyer, P.v.R. (Eds.) Stable Carbocation Chemistry; John Wiley and Sons: New York, NY, USA, 1997. [Google Scholar]
- Olah, G.A.; Prakash, G.K.S. (Eds.) Carbocation Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Stoyanov, E.S.; Gabriel, P.G. tert-Butyl Carbocation in Condensed Phases: Stabilization via Hyperconjugation, Polarization, and Hydrogen Bonding. J. Phys. Chem. A 2015, 119, 8619–8629. [Google Scholar] [CrossRef] [PubMed]
- Hanack, M. Mechanistic and preparative aspects of vinyl cation chemistry. Angew. Chem. Int. Ed. 1978, 17, 333–341. [Google Scholar] [CrossRef]
- Radom, L.; Hariharan, P.C.; Pople, J.A.; Schleyer, P.R. Molecular orbital theory of the electronic structure of organic compounds. XIX. Geometries and energies of C3H5 cations. Energy relations among allyl, vinyl, and cyclopropyl cations. J. Am. Chem. Soc. 1973, 95, 6531–6544. [Google Scholar] [CrossRef]
- Stang, P.J.; Rappoport, Z. Dicoordinated Carbocations; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Müller, T.; Juhasz, M.; Reed, C.A. The X-ray structure of a vinyl cation. Angew. Chem. Int. Ed. 2004, 43, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, R.J.; McNeil, A.J.; Thomas, Q.A.; Andrews, M.N. Primary Vinyl Cations in Solution: Kinetics and Products of β, β-Disubstituted Alkenyl(aryl)iodonium Triflate Fragmentations. J. Am. Chem. Soc. 1999, 121, 7437–7438. [Google Scholar] [CrossRef]
- Stang, P.J.; Rappaport, Z.; Hanack, M.; Subramanian, L.R. (Eds.) Vinyl Cations; Academic Press: San Diego, CA, USA, 1979. [Google Scholar]
- Siehl, H.-U.; Müller, T.; Gauss, J. NMR Spectroscopic and quantum chemical characterization of the (E )− and (Z )− isomers of the penta-1,3-dienyl-2-cation. J. Phys. Org. Chem. 2003, 16, 577–581. [Google Scholar] [CrossRef]
- Rablen, P.; Perry-Freer, N.A. How the arrangement of alkyl substituents affects the stability of delocalized carbocations. J. Org. Chem. 2018, 83, 4024–4033. [Google Scholar] [CrossRef] [PubMed]
- Cunji, A.; Rodriqyez, C.F.; Lien, M.H.; Hopkinson, A.C. The C4H5+ Potential Energy Surface. Structure, Relative Energies, and Enthalpies of Formation of Isomers of C4H5+. J. Org. Chem. 1996, 61, 5212–5220. [Google Scholar] [CrossRef]
- Byrne, P.A.; Kobayashi, S.; Wrthwein, E.-U.; Ammer, J.; Mayr, H. Why Are Vinyl Cations Sluggish Electrophiles? J. Am. Chem. Soc. 2017, 139, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, E.S.; Stoyanova, I.V. The Mechanism of High Reactivity of Benzyl Carbocation, C6H5CH2+, during Interaction with Benzene. Chem. Sel. 2020, 5, 9277–9280. [Google Scholar]
- Niggemann, M.; Gao, S. Are Vinyl Cations Finally Coming of Age? Angew. Chem. Int. Ed. 2018, 57, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P. Carbocation Chemistry; Elsevier: Amsterdam, The Netherlands, 1985; p. 173. [Google Scholar]
- Buzek, P.; Schleyer, P.R.; Vančik, H.; Mihalic, Z.; Gauss, J. Generation of the Parent Allyl Cation in a Superacid Cryogenic Matrix. Angew. Chem. Int. Ed. 1994, 33, 448–451. [Google Scholar] [CrossRef]
- Mišić, V.; Piech, K.; Bally, T. Carbocations Generated under Stable Conditions by Ionization of Matrix-Isolated Radicals: The Allyl and Benzyl Cations. J. Am. Chem. Soc. 2013, 135, 8625–8631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoyanov, E.S.; Bagryanskaya, I.Y.; Stoyanova, I.V. Unsaturated Vinyl-Type Carbocation [(CH3)2C=CH]+ in Its Carborane Salts. ACS Omega 2021, 6, 15834–15843. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, E.S.; Bagryanskaya, I.Y.; Stoyanova, I.V. Isomers of the Allyl Carbocation C3H5+ in Solid Salts: Infrared Spectra and Structures. ACS Omega 2021, 6, 23691–23699. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, E.S.; Bagryanskaya, I.Y.; Stoyanova, I.V. IR-Spectroscopic and X-ray- Structural Study of Vinyl-Type Carbocations in Their Carborane Salts. ACS Omega 2022, 7, 27560–27572. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.A. Carborane acids. New “strong yet gentle” acids for organic and inorganic chemistry. Chem. Commun. 2005, 13, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoyanov, E.S. Stabilization of Saturated Carbocations in Condensed Phases. J. Phys. Chem. A 2017, 121, 9638–9644. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, E.S.; Nizovtsev, A.S. Stabilization of carbocations CH3+, C2H5+, i-C3H7+, tert-Bu+, and cyclo-pentyl+ in solid phases: Experimental data versus calculations. Phys. Chem. Chem. Phys. 2017, 19, 7270–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoyanov, E.S.; Stoyanova, I.V. The Chloronium Cation, (C2H3)2Cl+, and Unsaturated C4-Carbocations with C=C and C≡C Bonds in Their Solid Salts and in Solutions: An H1/C13 NMR and Infrared Spectroscopic Study. Int. J. Mol. Sci. 2022, 23, 9111. [Google Scholar] [CrossRef]
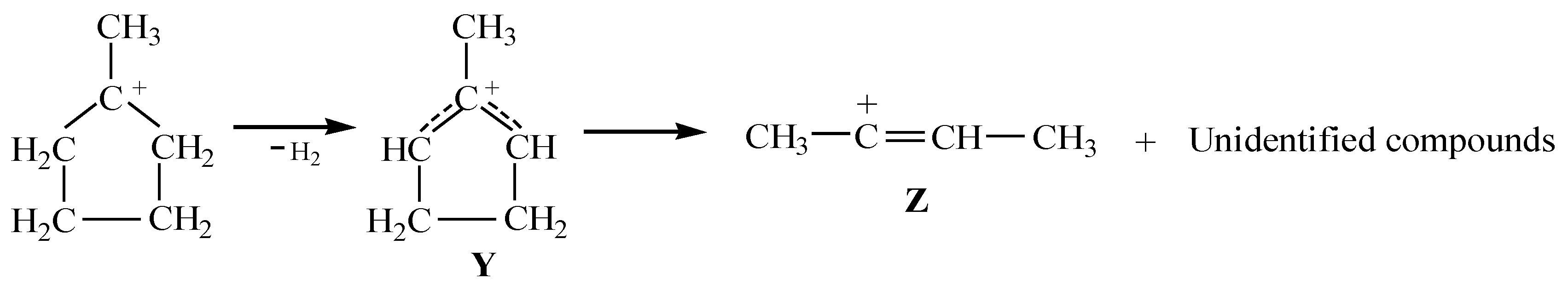
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoyanov, E.S.; Stoyanova, I.V. Spontaneous Transition of Alkyl Carbocations to Unsaturated Vinyl-Type Carbocations in Organic Solutions. Int. J. Mol. Sci. 2023, 24, 1802. https://doi.org/10.3390/ijms24021802
Stoyanov ES, Stoyanova IV. Spontaneous Transition of Alkyl Carbocations to Unsaturated Vinyl-Type Carbocations in Organic Solutions. International Journal of Molecular Sciences. 2023; 24(2):1802. https://doi.org/10.3390/ijms24021802
Chicago/Turabian StyleStoyanov, Evgenii S., and Irina V. Stoyanova. 2023. "Spontaneous Transition of Alkyl Carbocations to Unsaturated Vinyl-Type Carbocations in Organic Solutions" International Journal of Molecular Sciences 24, no. 2: 1802. https://doi.org/10.3390/ijms24021802
APA StyleStoyanov, E. S., & Stoyanova, I. V. (2023). Spontaneous Transition of Alkyl Carbocations to Unsaturated Vinyl-Type Carbocations in Organic Solutions. International Journal of Molecular Sciences, 24(2), 1802. https://doi.org/10.3390/ijms24021802