
Druggable Biomarkers Altered in Clear Cell Renal Cell Carcinoma: Strategy for the Development of Mechanism-Based Combination Therapy

Abstract
:1. Introduction
- (1)
- (2)
- (3)
- (4)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Markers | Sarc.ccRCC | Nno-Sarc ccRCC |
|---|---|---|---|
| [27] | PD-L1/TIL | 90% | 62% |
| [27] | PD-L1 | 54% | 17% |
| [27] | PD1/PD-L1 | 50% | 3% |
| [11] | HIF1 | 83% | 67% |
| [11] | HIF1/2 | 50% not expressed | 32% |
| [28] | miRs-210/-155 | over expressed | over expressed |
| [29] | Nrf2 | not reported | 78% |
2. Clear Cell Renal Cell Carcinoma
3. Druggable Targets Altered in ccRCC
3.1. MicroRNAs-210/-155 (miRs) and Hypoxia-Inducible Factor-1α and -2α (HIFS)
3.2. Transcription Factor Nrf2
3.3. Transforming Growth Factor-Beta (TGF-β)
3.4. P-Glycoprotein (Pgp)
4. Modulators of the Proposed Druggable Targets
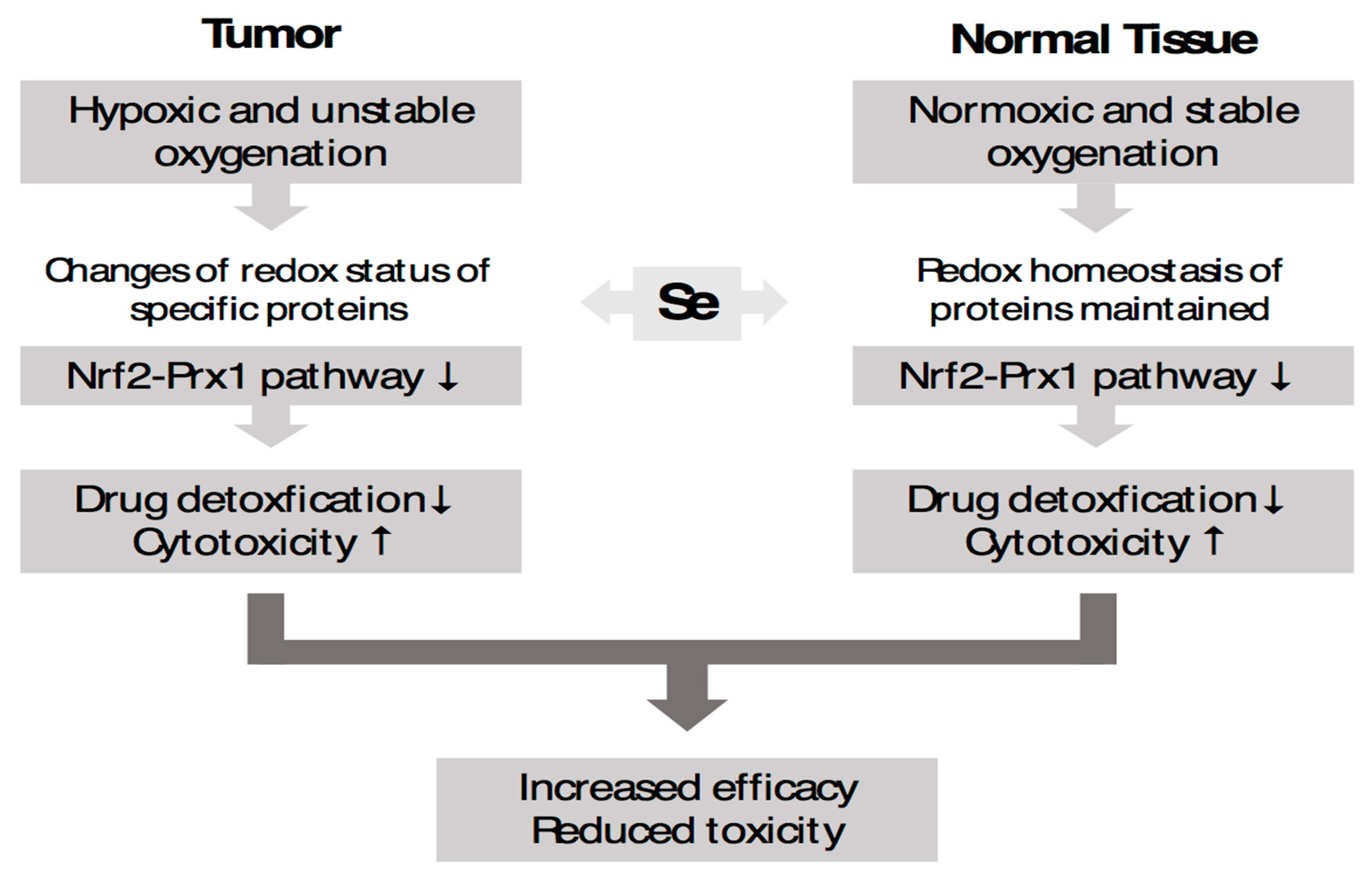
4.1. Selenium-Containing Molecules
4.2. Topotecan
5. Thymidine Phosphorylase, an Activator of 5-Flourouracil Pro-Drugs
6. Discussion
7. Take Home Message
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buonerba, C.; Dolce, P.; Iaccarino, S.; Scafuri, L.; Verde, A.; Costabile, F.; Pagliuca, M.; Morra, R.; Riccio, V.; Ribera, D.; et al. Outcomes Associated with First-Line anti-PD-1/PD-L1 agents vs. Sunitinib in Patients with Sarcomatoid Renal Cell Carcinoma: A Systematic Review and Meta-Analysis. Cancers 2020, 12, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T. Treatment Choices for Front-line Metastatic Clear Cell Renal Cancer. Eur. Urol. 2020, 77, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6, a027094. [Google Scholar] [CrossRef] [Green Version]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2014, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Treatment of kidney cancer. Cancer 2009, 115, 2262–2272. [Google Scholar] [CrossRef]
- Gao, W.; Li, W.; Xiao, T.; Liu, X.S.; Kaelin, W.G., Jr. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL−/−clear cell renal carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogner, A.; Kraus, H.; Jandrig, B.; Kasim, M.; Jandrig, B.; Kasim, M.; Fuller, T.F.; Schostak, M.; Erbersdobler, A.; Patzak, A.; et al. BRM1 and VHL Expression Correlate in Human Clear Cell Renal Cell Carcinoma with Differential Association with Patient’s Overall Survival. In Urologic Oncology; Springer: Cham, Switzerland, 2018; Volume 36. [Google Scholar]
- Brugarolas, J. PBRM1 and BAP1 as Novel Targets for Renal Cell Carcinoma. Cancer J. 2013, 19, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Ku, J.H.; Park, Y.H.; Myung, J.K.; Moon, J.K.; Kwak, C.; Kim, H.H. Expression of hypoxia inducible factor-1α and 2α in conventional renal cell carcinoma with or without sarcomatoid differentiation. Urol. Oncol. 2009, 29, 731–737. [Google Scholar] [CrossRef]
- Tóth, K.; Chintala, S.; Rustum, Y.M. Constitutive Expression of HIF-α Plays a Major Role in Generation of Clear-cell Phenotype in Human Primary and Metastatic Renal Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 642–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoefflin, R.; Harlander, S.; Schafer, S.; Metzger, P.; Kuo, F.; Schönenberger, D.; Adlesic, M.; Peighambari, A.; Seidel, P.; Chen, C.Y.; et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal carcinoma in mice. Nat. Commun. 2020, 11, 4111. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, C. Differential Roles of Hypoxia- Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Raval, R.R.; Lau, K.W.; Tran, M.G.B.; Sowter, H.M.; Mandriota, S.J.; Li, J.-L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting Properties of Hypoxia-Inducible Factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-Associated Renal Cell Carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, Y.; Song, Y.; Bu, R.; Yin, B.; Fei, X.; Guo, Q.; Wu, B. MicroRNAs in renal cell carcinoma: A systematic review of clinical implications (Review). Oncol. Rep. 2015, 33, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Rustum, Y.M.; Chintala, S.; Durrani, F.A.; Bhattacharya, A. Non-Coding Micro RNAs and Hypoxia-Inducible Factors Are Selenium Targets for Development of a Mechanism-Based Combination Strategy in Clear-Cell Renal Cell Carcinoma—Bench-to-Bedside Therapy. Int. J. Mol. Sci. 2018, 19, 3378. [Google Scholar] [CrossRef] [Green Version]
- Mytsyk, Y.; Dosenko, V.; Skrzypczyk, M.A.; Borys, Y.; Diychuk, Y.; Kucher, A.; Kowalskyy, V.; Pasichnyk, S.; Mytsyk, O.; Manyuk, L. Potential clinical application of micrRNAs as biomarker for renal cell carcinoma. Cent. Eur. J. Urol. 2018, 71, 295–303. [Google Scholar]
- Shiomi, E.; Sugai, T.; Ishida, K.; Osakabe, M.; Tsuyukubo, T.; Kato, Y.; Takata, R.; Obara, W. Analysis of Expression Patterns of MicroRNAs That Are Closely Associated With Renal Carcinogenesis. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Redova, M.; Svoboda, M.; Slaby, O. MicroRNAs and their target gene networks in renal cell carcinoma. Biochem. Biophys. Res. Commun. 2011, 405, 153–156. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Y.; Chang, D.W.; Lin, S.-H.; Huang, M.; Lin, S.H.; Huang, M.; Tannir, N.M.; Matin, S.; Karam, J.A.; et al. Global and Targeted miRNA Expression Profiling in Clear Cell Renal Cell Carcinoma Tissues Potentially Links miR-155-5p and miR-210- 3p to both Tumorigenesis and Recurrence. Am. J. Pathol. 2018, 188, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhang, L.; Brett-Morris, A.; Agujila, A.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.R.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Ruan, H.; Song, Z.S.; Cao, Q.; Bao, L.; Liu, D.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; et al. PLIN3 is upregulated and correlates with poor prognosis in clear cell renal cell carcinoma. Urol. Oncol. 2018, 36, 343.e9–343.e19. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Ruan, H.; Wang, K.; Song, Z.; Bao, L.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; Tong, J.; et al. Overexpression of PLIN2 is a prognostic marker and attenuates tumor progression in clear cell renal cell carcinoma. Int. J. Oncol. 2018, 53, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, E.; Osaki, M.; Kase, S.; Adachi, H.; dachi, H.; Kaibara, N.; Ito, H. Thymidine phosphorylase expression causes both the increase of intertumoral microvessels and decrease of apoptosis in human esophageal carcinomas. Pathol. Int. 2008, 51, 158–164. [Google Scholar] [CrossRef]
- Joseph, R.W.; Millis, S.Z.; Carballido, E.M.; Bryant, D.; Gatalica, Z.; Reddy, S.; Bryce, A.H.; Vogelzang, N.J.; Stanton, M.L.; Castle, E.P.; et al. PD-1 and PD-L1 Expression in Renal Cell Carcinoma with Sarcomatoid Differentiation. Cancer Immunol. Res. 2015, 3, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Tian, D.; Zhang, B.; Zhang, Y.; Yan, D.; Wu, S. Overexpression of miR-155 in clear-cell renal cell carcinoma and its oncogenic effect through targeting FOXO3a. Exp. Ther. Med. 2017, 13, 2286–2292. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Wu, Y.; Zhao, P.; Weng, W.; Ye, M.; Sun, H.; Xu, M.; Wang, C. The Nrf2/HO-1 axis can be a prognostic factor in clear cell renal cell carcinoma. Cancer Manag. Res. 2019, ume 11, 1221–1230. [Google Scholar] [CrossRef] [Green Version]
- Facchini, G.; Rossetti, S.; Berretta, M.; Cavaliere, C.; Scagliarini, S.; Vitale, M.G.; Ciccarese, C.; Di Lorenzo, G.; Palesandro, E.; Conteduca, V.; et al. Second line therapy with axitinib after only prior sunitinib in metastatic renal cell cancer: Italian multicenter real world SAX study final results. J. Transl. Med. 2019, 17, 1–11. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomized phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Kammerer-Jacquet, S.-F.; Crouzet, L.; Brunot, A.; Dagher, J.; Pladys, A.; Edeline, J.; Laguerre, B.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; et al. Independent association of PD-L1 expression with noninactivated VHL clear cell renal cell carcinoma-A finding with therapeutic potential. Int. J. Cancer 2016, 140, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kim, T.B.; Peng, B.; Karam, J.; Creighton, C.; Joon, A.; Kawakami, F.; Trevisan, P.; Jonasch, E.; Chow, C.-W.; et al. Sarcomatoid Renal Cell Carcinoma Has a Distinct Molecular Pathogenesis, Driver Mutation Profile, and Transcriptional Landscape. Clin. Cancer Res. 2017, 23, 6686–6696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, M.; Zhao, S.; Said, J.W.; Merino, M.J.; Adeniran, A.J.; Xie, Z.; Nawaf, C.B.; Choi, J.; Belldegrun, A.S.; Pantuck, A.J.; et al. Genomic characterization of sarcomatoid transformation in clear cell renal cell carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 2170–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, K.A.; Gupta, S.; Tichoo, S.K.; Chan, T.A.; Russo, P.; Motzer, R.J.; karam, J.A.; Hakima, A. Sarcomatoid renal cell carci-noma: Biology, natural history and management. Nat. Rev. Urol. 2020, 17, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Bailey, D. Tumor vascularization: A druggable target. Curr. Opin. Pharmacol. 2009, 9, 96–100. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chen, T.-H.; Huang, Y.-M.; Wei, P.-L.; Lin, J.-C. Involvement of microRNA in Solid Cancer: Role and Regulatory Mechanisms. Biomedicines 2021, 9, 343. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, L.; Li, Y.; Huang, C.; Zeng, L.; Yang, J. Differential microRNA expression in renal cell carcinoma. Oncol. Lett. 2013, 6, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Heidegger, I.; Pircher, A.; Pichler, R. Targeting the Tumor Microenvironment in Renal Cell Cancer Biology and Therapy. Front. Oncol. 2019, 9, 490. [Google Scholar] [CrossRef] [Green Version]
- Schanza, L.-M.; Seles, M.; Stotz, M.; Fosselteder, J.; Hutterer, G.C.; Pichler, M.; Stiegelbauer, V. MicroRNAs Associated with Von Hippel–Lindau Pathway in Renal Cell Carcinoma: A Comprehensive Review. Int. J. Mol. Sci. 2017, 18, 2495. [Google Scholar] [CrossRef] [Green Version]
- An, X.; Sarmiento, C.; Tan, T.; Zhu, H. Regulation of multidrug resistance by microRNAs in anti-cancer therapy. Acta Pharm. Sin. B 2017, 7, 38–51. [Google Scholar] [CrossRef]
- Ma, J.; Dong, C.; Ji, C. MicroRNA and drug resistance. Cancer Gene Ther. 2010, 17, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, F.P.; Sparaneo, A.; Muscarella, L.A. NRF2 Regulation by Noncoding RNAs in Cancers: The Present Knowledge and the Way Forward. Cancers 2020, 12, 3621. [Google Scholar] [CrossRef] [PubMed]
- Puissegur, M.-P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is over expressed in late-stage Lung cancer and mediate mitochondrial alterations associated with modulation of HIE-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.; Zhao, L.; Sun, H.; Pan, K.; Sun, H.; Zhang, Z.; Zhou, M.; Cao, G.; Wang, M. GPD1L is down regulated by three- mi-croRNA signature in Pancreatic Cancer. Transl. Cancer Res. 2017, 6, 106. [Google Scholar] [CrossRef]
- Kelly, T.J.; Souza, A.L.; Clish, C.B.; Puigserver, P. A Hypoxia-Induced Positive Feedback Loop Promotes Hypoxia-Inducible Factor 1α Stability through miR-210 Suppression of Glycerol-3-Phosphate Dehydrogenase 1-Like. Mol. Cell. Biol. 2011, 31, 2696–2706. [Google Scholar] [CrossRef] [Green Version]
- Kulshreshtha, R.; Ferracin, M.; Wojcik, S.; Garzon, R.; Alder, H.; Agosto-Perez, F.J.; Davuluri, R.; Liu, C.G.; Croce, C.M.; Negrini, M.; et al. A microRNA signature of hypoxia. Mol. Cell. Biol. 2007, 27, 1859–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorko, M.; Pacik, D.; Wasserbauer, R.; Juracek, J.; Varga, G.; Ghazal, M.; Nussir, M.I. MicroRNAs in the Pathogenesis of Renal Cell Carcinoma and Their Diagnostic and Prognostic Utility as Cancer Biomarkers. Int. J. Biol. Markers 2016, 31, 26–37. [Google Scholar] [CrossRef]
- Braga, E.A.; Fridman, M.V.; Loginov, V.I.; Dmitriev, A.A.; Morozov, S.G. Molecular Mechanisms in Clear Cell Renal Cell Carcinoma: Role of miRNAs and Hypermethylated miRNA Genes in Crucial Oncogenic Pathways and Processes. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Van Peer, G.; Mets, E.; Claeys, S.; De Punt, I.; Lefever, S.; Ongenaert, M.; Rondou, P.; Speleman, F.; Mestdagh, P.; Vandesompele, J. A high-throughput 3′ UTR reporter screening identifies microRNA interactomes of cancer genes. PLoS ONE 2018, 13, e0194017. [Google Scholar] [CrossRef] [Green Version]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [Green Version]
- Zakharia, Y.; Bhattacharya, A.; Rustum, Y.M. Selenium targets resistance biomarkers enhancing efficacy while reducing tox-icity of anti-cancer drugs: Preclinical and clinical development. Oncotarget 2018, 9, 10765–10783. [Google Scholar] [CrossRef] [PubMed]
- Chintala, S.; Najrana, T.; Toth, K.; Cao, S.; Durrani, F.A.; Pili, R.; Rustum, Y.M. Prolyl hydroxylase 2 dependent and Von-Hippel-Lindau independent degradation of hypoxia-inducible factor 1 and 2 alpha by selenium in clear cell renal cell carcinoma leads to tumor growth inhibition. BMC Cancer 2012, 12, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chintala, S.; Toth, K.; Cao, S.; Durrani, F.A.; Vaughan, M.M.; Jensen, R.L.; Rustum, Y.M. Se-methylselenocysteine sensitizes hypoxic tumor cells by targeting hypoxia inducible factor1α. Cancer Chemother. Pharm. 2010, 66, 899–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranni, F.; Chintala, S.; Toth, K.; Rustum, Y.M. Mechanism-based drug combination targeting HIF2α and VEGF. Trends Cell Mol. Biol. 2015, 10, 2561–2569. [Google Scholar]
- Cao, S.; Durrani, F.A.; Rustum, Y.M. Selective modulation of the therapeutic efficacy of anticancer drugs by selenium con-taining compounds against human tumor xenografts. Clin. Cancer Res. 2004, 10, 256. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Seshadri, M.; Oven, S.D.; Tóth, K.; Vaughan, M.M.; Rustum, Y.M. Tumor Vascular Maturation and Improved Drug Delivery Induced by Methylselenocysteine Leads to Therapeutic Synergy with Anticancer Drugs. Clin. Cancer Res. 2008, 14, 3926–3932. [Google Scholar] [CrossRef] [Green Version]
- Ooi, A.; Furge, K.A. Fumarate hydratase inactivation in renal tumors: HIF1 alpha, NRF2, and “cryptic targets” of transcription factors. Chin. J. Cancer 2012, 31, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Cornejo, K.M.; Lu, M.; Yang, P.; Wu, S.; Cai, C.; Zhong, W.-D.; Olumi, A.; Young, R.H.; Wu, C.-L. Succinate dehydrogenase B: A new prognostic biomarker in clear cell renal cell carcinoma. Hum. Pathol. 2015, 46, 820–826. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Mu, X.; You, Q. Succinate: An initiator in tumorigenesis and progression. Oncotarget 2017, 8, 53819–53828. [Google Scholar] [CrossRef] [Green Version]
- Bruning, U.; Cerone, L.; Neufeld, Z.; Fitzpatrick, S.F.; Fitzpatrick, S.F.; Cheong, A.; Scholz, C.C.; Simpson, D.A.; Leonard, M.O.; Tambuwala, M.M.; et al. MicroRNA-155 promote resolution of hypoxia-inducible factor 1 alpha activity during prolonged hypoxia. Mol. Cell Biol. 2011, 19, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.-D.; Chang, S. Development of Novel Therapeutic Agents by Inhibition of Oncogenic MicroRNAs. Int. J. Mol. Sci. 2018, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, T.H.; Li, Q.; Ma, H.W.; Key, J.; Zhang, L.; Chen, R.; Garcia, J.A.; Naidoo, J.; Longgood, J.; Frantz, D.E.; et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat. Chem. Biol. 2013, 9, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, T.H.; Tomchick, D.R.; Machius, M.; Guo, Y.; Bruick, R.K.; Gardner, K.H. Artificial ligand binding within the HIF2α PAS-B domain of the HIF2 transcription factor. Proc. Natl. Acad. Sci. USA 2009, 106, 450–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezu, M.; Suzuki, N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int. J. Mol. Sci. 2020, 21, 2951. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Yuki, H.; Kamai, T.; Murakami, S.; Higasi, S.; Narimatsu, T.; Higashi, S.; Narimatsu, T.; Kambara, T.; Betsunoh, H.; Abe, H. Increased Nrf2 expression by renal cell carcinoma is associated with postoperative chronic kidney disease and an unfavorable prognosis. Oncotarget 2018, 9, 28351–28352. [Google Scholar] [CrossRef]
- Clerici, S.; Boletta, A. Role of the KEAP1-NRF2 Axis in Renal Cell Carcinoma. Cancers 2020, 12, 3458. [Google Scholar] [CrossRef]
- Bocci, F.; Tripathi, S.C.; Mercedes, S.A.V.; George, J.T.; Casabar, J.P.; Wong, P.K.; Hanash, S.M.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr. Biol. 2019, 11, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-C.; Baek, S.-H.; Bogner, P.N.; Ip, C.; Rustum, y.; Fakih, M.; Ramnath, N.; Park, Y.-M. Targeting the Nrf2-Prx1 pathway with selenium to enhance the efficacy and selectivity of cancer therapy. J. Cancer Mol. 2007, 3, 37–43. [Google Scholar]
- Müller, M.; Banning, A.; Brigelius-Flohé, R.; Kipp, A. Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 2010, 5, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tindell, R.; Wall, S.B.; Li, Q.; Li, R.; Dunigan, K.; Wood, R.; Tipple, T.E. Selenium supplementation of lung epithelial cells enhances nuclear factor E2-related factor 2 (Nrf2) activation following thioredoxin reductase inhibition. Redox Biol. 2018, 19, 331–338. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Reszka, E.; Wieczorek, E.; Jablonska, E.; Janasik, B.; Fendler, W.; Wasowicz, W. Association between plasma selenium level and NRF2 target genes expression in humans. J. Trace Elements Med. Biol. 2015, 30, 102–106. [Google Scholar] [CrossRef]
- Bilim, V.; Ougolkov, A.; Yuuki, K.; Naito, S.; Kawazoe, H.; Muto, A.; Oya, M.; Billadeau, D.; Motoyama, T.; Tomita, Y. Glycogen synthase kinase-3: A new therapeutic target in renal cell carcinoma. Br. J. Cancer 2009, 101, 2005–2014. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Xiaoming, X.; Yong, L.; Yuanfa, F.; Rui, Z.; Weide, Z. Expression and clinical significance of GPD1L in clear cell renal carcinoma. J. New Med. 2020, 51, 127–132. [Google Scholar] [CrossRef]
- Sudarshan, S.; Sourbier, C.; Kong, H.S.; Block, K.; Valera Romero, V.A.; Yang, Y.; Galindo, C.; Mollapour, M.; Scroggins, B.; Goode, N.; et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol. Cell Biol. 2009, 29, 4080–4090. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Sun, Q.; Yang, H.; Zheng, J. SDHB Suppresses the Tumorigenesis and Development of ccRCC by Inhibiting Glycolysis. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Zhou, J.; Dabiri, Y.; Gama-Brambila, R.A.; Ghafoory, S.; Altinbay, M.; Mehrabi, A.; Golriz, M.; Blagojevic, B.; Reuter, S.; Han, K.; et al. pVHL-mediated SMAD3 degradation suppresses TGF-β signaling. J. Cell Biol. 2021, 221. [Google Scholar] [CrossRef]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallikarjuna, P. The Role of Transforming Growth Factor-B Signaling and Hypoxia-Inducible Factors in Renal Cell Carcinoma. Ph.D. Thesis, Umea University, Umea, Sweden, 2019. [Google Scholar]
- Mallikarjuna, P.; Sitaram, R.T.; Aripaka, K.; Ljungberg, B.; Landström, M. Interactions between TGF-β type I receptor and hypoxia-inducible factor-α mediates a synergistic crosstalk leading to poor prognosis for patients with clear cell renal cell carcinoma. Cell Cycle 2019, 18, 2141–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zonneville, J.; Safina, A.; Truskinovsky, A.M.; Arteaga, C.L.; Bakin, A.V. TGF-β signaling promotes tumor vasculature by enhancing the pericyte-endothelium association. BMC Cancer 2018, 18, 670. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef] [Green Version]
- Hagemann-Jensen, M.; Uhlenbrock, F.; Kehlet, S.; Andresen, L.; Gabel-Jensen, C.; Ellgaard, L.; Gammelgaard, B.; Skov, S. The Selenium Metabolite Methylselenol Regulates the Expression of Ligands That Trigger Immune Activation through the Lymphocyte Receptor NKG2D. J. Biol. Chem. 2014, 289, 31576–31590. [Google Scholar] [CrossRef] [Green Version]
- Chen, W. A potential treatment of COVID-19 with TGF-β blockade. Int. J. Biol. Sci. 2020, 16, 1954–1955. [Google Scholar] [CrossRef]
- Khatiwada, S.; Subedi, A. A Mechanistic Link Between Selenium and Coronavirus Disease 2019 (COVID-19). Curr. Nutr. Rep. 2021, 10, 125–136. [Google Scholar] [CrossRef]
- Sell, K.; Barth, P.J.; Moll, R.; Thomas, M.A.; Zimmer, N.; Oplesch, E.; Gudo, M.; Schrader, M.; Hofmann, R.; Schrader, A.J. Localization of FOXP3-positive cells in renal cell carcinoma. Tumor Biol. 2011, 33, 507–513. [Google Scholar] [CrossRef]
- Naito, S.; Sakamoto, N.; Kotoh, S.; Goto, K.; Matsumoto, T.; Kumazawa, J. Expression of P- glycoprotein and multidrug re-sistance in renal cell carcinoma. Eur. Urol. 1993, 24, 156–160. [Google Scholar]
- Bak, J.M.; Efferth, T.; Mickisch, G.; Mattern, J.; Volm, M. Detection of Drug Resistance and P-Glycoprotein in Human Renal Cell Carcinomas. Eur. Urol. 1990, 17, 72–75. [Google Scholar] [CrossRef]
- Mignogna, C.; Staibano, S.; Altieri, V.; De Rosa, G.; Pannone, G.; Santoro, A.; Zamparese, R.; D’Armiento, M.; Rocchetti, R.; Mezza, E.; et al. Prognostic significance of multidrug-resistance protein (MDR-1) in renal clear cell carcinomas: A five year follow-up analysis. BMC Cancer 2006, 6, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef]
- Shibayama, Y.; Nakano, K.; Maeda, H.; Taguchi, M.; Ikeda, R.; Sugawara, M.; Iseki, K.; Takeda, Y.; Yamada, K. Multidrug Resistance Protein 2 Implicates Anticancer Drug-Resistance to Sorafenib. Biol. Pharm. Bull. 2011, 34, 433–435. [Google Scholar] [CrossRef] [Green Version]
- Bielecka, Z.F.; Czarnecka, A.M.; Solarek, W.; Kornakiewicz, A. Mechanisms of Acquired Resistance to Tyrosine Kinase Inhib-itors in Clear-Cell Renal Cell Carcinoma (ccRCC). Curr. Signal Transduct. Ther. 2013, 8, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Jedeszko, C.; Paez-Ribes, M.; Di Desidero, T.; Man, S.; Lee, C.R.; Xu, P.; Bjarnason, G.A.; Bocci, G.; Kerbel, R.S. Postsurgical adjuvant or metastatic renal cell carcinoma therapy models reveal potent antitumor activity of metronomic oral topotecan with pazopanib. Sci. Transl. Med. 2015, 7, 282ra50-282ra50. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Infante, J.R.; Lam, E.T. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Fac-tor-2alpha Antagonist in Patients with Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Raja, R.; Kuziora, M.; Brohawn, P.Z.; Higgs, B.W.; Gupta, A.; Dennis, P.A.; Ranade, K. Early reduction in ctDNA predicts sur-vival in patients with lung and bladder cancer treated with durvalumab. Clin. Cancer Res. 2018, 24, 6212–6222. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.M.; Choueiri, T.K.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. The oral HIF-2 α inhibitor MK-6482 in patients with advanced clear cell renal cell carcinoma (RCC): Updated follow-up of a phase I/II study. J. Clin. Oncol. 2021, 39, 273. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, L.; Li, S.; Chen, F.; Au-Yeung, K.K.-W.; Shi, C. MicroRNA as an Important Target for Anticancer Drug Development. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Ebert, R.; Ulmer, M.; Zeck, S.; Meissner-Weigl, J.; Schneider, D.; Stopper, H.; Schupp, N.; Kassem, M.; Jakob, F. Selenium sup-plementation restores the antioxidative capacity and prevents cell damage in bone marrow stromal cells in vitro. Stem Cells 2006, 24, 1226–1235. [Google Scholar] [CrossRef]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, R.F. Selenium, an Antioxidant Nutrient. Nutr. Clin. Care 2002, 5, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A. Methylselenocysteine—A promising antiangiogenic agent for overcoming drug delivery barriers in solid malignancies for therapeutic synergy with anticancer drugs. Expert Opin. Drug Deliv. 2011, 8, 749–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azrak, R.G.; Cao, S.; Durrani, F.A.; Toth, K.; Bhattacharya, A.; Rustum, Y.M. Augmented therapeutic efficacy of irinotecan is associated with enhanced drug accumulation. Cancer Lett. 2011, 311, 219–229. [Google Scholar] [CrossRef]
- Rustum, Y.M.; Tóth, K.; Seshadri, M.; Sen, A.; Durrani, F.A.; Stott, E.; Morrison, C.D.; Cao, S.; Bhattacharya, A. Architectural Heterogeneity in Tumors Caused by Differentiation Alters Intratumoral Drug Distribution and Affects Therapeutic Synergy of Antiangiogenic Organoselenium Compound. J. Oncol. 2010, 2010, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippman, S.M.; Goodman, P.J.; Klein, E.A.; Parnes, H.L.; Thompson, I.M.; Kristal, A.; Santella, R.M.; Probstfield, J.L.; Moinpour, C.M.; Albanes, D.; et al. Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JNCI: J. Natl. Cancer Inst. 2005, 97, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the Risk of Prostate Cancer: The selenium and vitamin e cancer prevention trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Farhood, B.; Mortezaee, K.; Motevaseli, E.; Mirtavoos-Mahyari, H.; Shabeeb, D.; Musa, A.E.; Sanikhani, N.S.; Najafi, M.; Ahmadi, A. Selenium as an adjuvant for modification of radiation response. J. Cell Biochem. 2019, 120, 18559–18571. [Google Scholar] [CrossRef]
- Lobb, R.J.; Jacobson, G.M.; Cursons, R.T.; Jameson, M.B. The Interaction of Selenium with Chemotherapy and Radiation on Normal and Malignant Human Mononuclear Blood Cells. Int. J. Mol. Sci. 2018, 19, 3167. [Google Scholar] [CrossRef] [Green Version]
- Fritz, H.; Kennedy, D.; Fergusson, D.; Fernandes, R.; Cooley, K.; Seely, A.; Sagar, S.; Wong, R.; Seely, D. Selenium and Lung Cancer: A Systematic Review and Meta Analysis. PLoS ONE 2011, 6, e26259. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, Z.; Cao, Y.; Lu, W.; Kuang, L.; Hua, D. Strategy for Highly Efficient Radioprotection by a Selenium-Containing Polymeric Drug with Low Toxicity and Long Circulation. ACS Appl. Mater. Interfaces 2020, 12, 44534–44540. [Google Scholar] [CrossRef] [PubMed]
- Zakharia, Y.; Sieren, J.; Reis, R.; Garje, R.; Born, J.; Rajput, M.; Humble, R.; Bellizzi, A.; Rustum, Y.M. Potential Role of Sele-no-L-Methionine (SLM) in the Stabilization of Tumor Vasculature and Enhanced Efficacy of Axitinib in Previously Treated Patients with Advanced Clear Cell Renal Cell Carcinoma (ccRCC). In Proceedings of the Kidney Cancer Research Summit, Philadelphia, PA, USA, 7–8 October 2021. [Google Scholar]
- Puppo, M.; Battaglia, F.; Ottaviano, C.; Delfino, S.; Ribatti, D.; Varesio, L.; Bosco, M.C. Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1α and -2α. Mol. Cancer Ther. 2008, 7, 1974–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapisarda, A.; Zalek, J.; Hollingshead, M.; Braunschweig, T.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Hewitt, S.M.; Shoemaker, R.H.; et al. Schedule-dependent inhibition of hypoxia- inducible factor-1alpha protein accumulation, an-giogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res. 2004, 64, 6845–6882. [Google Scholar] [CrossRef] [Green Version]
- Bernstock, J.D.; Ye, D.; Gessle, F.A.; Luca Peruzzotti-Jametti, L.; Gilbert, M.R.; Pommier, Y.; Pluchino, S.; Nikano, I.; Hallenbeck, J. Topotecan Decreases the Expression of Programmed Death-Ligand 1 in Glioblastoma Cell Lines; Implications for Immunotherapy. Matters 2017, 3. [Google Scholar] [CrossRef]
- Hashimoto, K.; Man, S.; Xu, P.; Cruz-Munoz, W.; Tang, T.; Kumar, R.; Kerbel, R.S. Potent Preclinical Impact of Metronomic Low-Dose Oral Topotecan Combined with the Antiangiogenic Drug Pazopanib for the Treatment of Ovarian Cancer. Mol. Cancer Ther. 2010, 9, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, T.; Mugiya, S.; Sugiyama, T.; Aoki, T.; Furuse, H.; Liu, H.; Hirano, Y.; Kai, F.; Ushiyama, T.; Ozono, S. High Levels of Thymidine Phosphorylase as an Independent Prognostic Factor in Renal Cell Carcinoma. Jpn. J. Clin. Oncol. 2006, 36, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Wang, L.; Chen, Y.; Zheng, X.; Wang, X. Poor Prognosis Associated with High Levels of Thymidine Phosphorylase and Thrombocytosis in Patients with Renal Cell Carcinoma. Urol. Int. 2016, 98, 162–168. [Google Scholar] [CrossRef]
- Oevermann, K.; Buer, J.; Hoffmann, R.; Franzke, A.; Schrader, A.; Patzelt, T.; Kirchner, H.; Atzpodien, J. Capecitabine in the treatment of metastatic renal cell carcinoma. Br. J. Cancer 2000, 83, 583–587. [Google Scholar] [CrossRef] [Green Version]
- Elamin, Y.Y.; Rafee, S.; Osman, N.; O’Byrne, K.J.; Gately, K. Thymidine Phosphorylase in Cancer; Enemy or Friend? Cancer Microenviron. 2016, 9, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Farha, M.; Vince, R.; Nallandhighal, S.; Stangl-Kremser, J.; Goldenthal, S.; Triner, D.; Morgan, T.M.; Palapattu, G.S.; Udager, A.M.; Salami, S.S. Characterization of the tumor immune microenvironment in clear cell renal cell carcinoma (ccRCC): Prognostic value and therapeutic implications of an M0-macrophage enriched subtype. J. Clin. Oncol. 2021, 39, 4572-4572. [Google Scholar] [CrossRef]
- Arreola, A.; Cowey, C.L.; Coloff, J.L.; Rathmell, J.C.; Rathmell, W.K. HIF1α and HIF2α Exert Distinct Nutrient Preferences in Renal Cells. PLoS ONE 2014, 9, e98705. [Google Scholar] [CrossRef] [Green Version]
- Branco-Price, C.; Zhang, N.; Schnelle, M.; Evans, C.; Katschinski, D.M.; Liao, D.; Ellies, L.; Johnson, R.S. Endothelial Cell HIF-1α and HIF-2α Differentially Regulate Metastatic Success. Cancer Cell 2012, 21, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidian, A.; Stedingk, K.; Munksgaard, M.; Mohlin, T.S.; Pahlman, S. Differential regulation of HIF-1α and HIF-2α in neuroblastoma: Estrogen-related receptor alpha (ERRα) regulates HIF2Atranscription and to poor outcome. Biochem. Biophys. Res. Commun. 2015, 461, 560–567. [Google Scholar] [CrossRef]
- Bombelli, S.; Torsello, B.; De Marco, S.; Lucarelli, G.; Cifola, I.; Grasselli, C.; Strada, G.; Bovo, G.; Perego, R.A.; Bianchi, C. 36-kDa Annexin A3 Isoform Negatively Modulates Lipid Storage in Clear Cell Renal Cell Carcinoma Cells. Am. J. Pathol. 2020, 190, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Meregalli, C.; Bombelli, S.; Di Stefano, V.; Salerno, F.; Torsello, B.; De Marco, S.; Bovo, G.; Cifola, I.; Mangano, E.; et al. The glucose and lipid metabolism reprogramming is grade-dependent in clear cell renal cell carcinoma primary cultures and is targetable to modulate cell viability and proliferation. Oncotarget 2017, 8, 113502–113515. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Loizzo, D.; Franzin, R.; Battaglia, S.; Ferro, M.; Cantiello, F.; Castellano, G.; Bettocchi, C.; Ditonno, P.; Battaglia, M. Metabolomic insights into pathophysiological mechanisms and biomarker discovery in clear cell renal cell carcinoma. Expert Rev. Mol. Diagn. 2019, 19, 397–407. [Google Scholar] [CrossRef]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Loizzo, D.; Ferro, M.; Stella, A.; Bizzoca, C.; Vincenti, L.; Pandolfo, S.D.; Autorino, R.; et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 14360. [Google Scholar] [CrossRef]
- De Marco, S.; Torsello, B.; Minutiello, E.; Morabito, I.; Grasselli, C.; Bombelli, S.; Zucchini, N.; Lucarelli, G.; Strada, G.; Perego, R.A.; et al. The cross-talk between Abl2 tyrosine kinase and TGFβ1 signalling modulates the invasion of clear cell Renal Cell Carcinoma cells. FEBS Lett. 2022. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Signorile, M.L.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated multi-omics characterization reveals a distinctive metabolic signature and the role of NDUFA4L2 in promoting angiogenesis, chemoresistance, and mitochondrial dysfunction in clear cell renal cell carcinoma. Aging 2018, 10, 3957–3985. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Loizzo, D.; di Meo, N.A.; Lasorsa, F.; Mastropasqua, M.; Maiorano, E.; Bizzoca, C.; Vincenti, L.; Battaglia, M.; et al. MUC1 Tissue Expression and Its Soluble Form CA15-3 Identify a Clear Cell Renal Cell Carcinoma with Distinct Metabolic Profile and Poor Clinical Outcome. Int. J. Mol. Sci. 2022, 23, 13968. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Rutigliano, M.; Ferro, M.; Giglio, A.; Intini, A.; Triggiano, F.; Palazzo, S.; Gigante, M.; Castellano, G.; Ranieri, E.; et al. Activation of the kynurenine pathway predicts poor outcome in patients with clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 461.e15–461.e27. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.R.; Kim, S.I.; Kim, S.J.; Cho, D.S. Prognostic nutritional index as a prognostic factor for renal cell carcinoma: A sys-tematic review and meta-analysis. PLoS ONE 2022, 17, e0271821. [Google Scholar] [CrossRef]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor Microenvironment Dynamics in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2019, 9, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Tamma, R.; Rutigliano, M.; Lucarelli, G.; Annese, T.; Ruggieri, S.; Cascardi, E.; Napoli, A.; Battaglia, M.; Ribatti, D. Microvascular density, macrophages, and mast cells in human clear cell renal carcinoma with and without bevacizumab treatment. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 355.e11–355.e19. [Google Scholar] [CrossRef]
- Zhang, J.; Saad, R.; Taylor, E.W.; Rayman, M.P. Selenium and selenoproteins in viral infection with potential relevance to COVID-19. Redox Biol. 2020, 37, 101715. [Google Scholar] [CrossRef]
- Moghaddam, A.; Heller, R.A.; Sun, Q.; Seelig, J.; Cherkezov, A.; Seibert, L.; Hackler, J.; Seemann, P.; Diegmann, J.; Pilz, M.; et al. Selenium Deficiency Is Associated with Mortality Risk from COVID-19. Nutrients 2020, 12, 2098. [Google Scholar] [CrossRef]
- Zhang, J.; Taylor, E.W.; Bennett, K.; Saad, R.; Rayman, M.P. Association between regional selenium status and reported outcome of COVID-19 cases in China. Am. J. Clin. Nutr. 2020, 111, 1297–1299. [Google Scholar] [CrossRef]
- Ferreira-Gomes, M.; Kruglov, A.; Durek, P.; Heinrich, F.; Tizian, C.; Heinz, G.A.; Pascual-Reguant, A.; Du, W.; Mothes, R.; Fan, C.; et al. SARS-CoV-2 in severe COVID-19 induces a TGF-β-dominated chronic immune response that does not target itself. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]

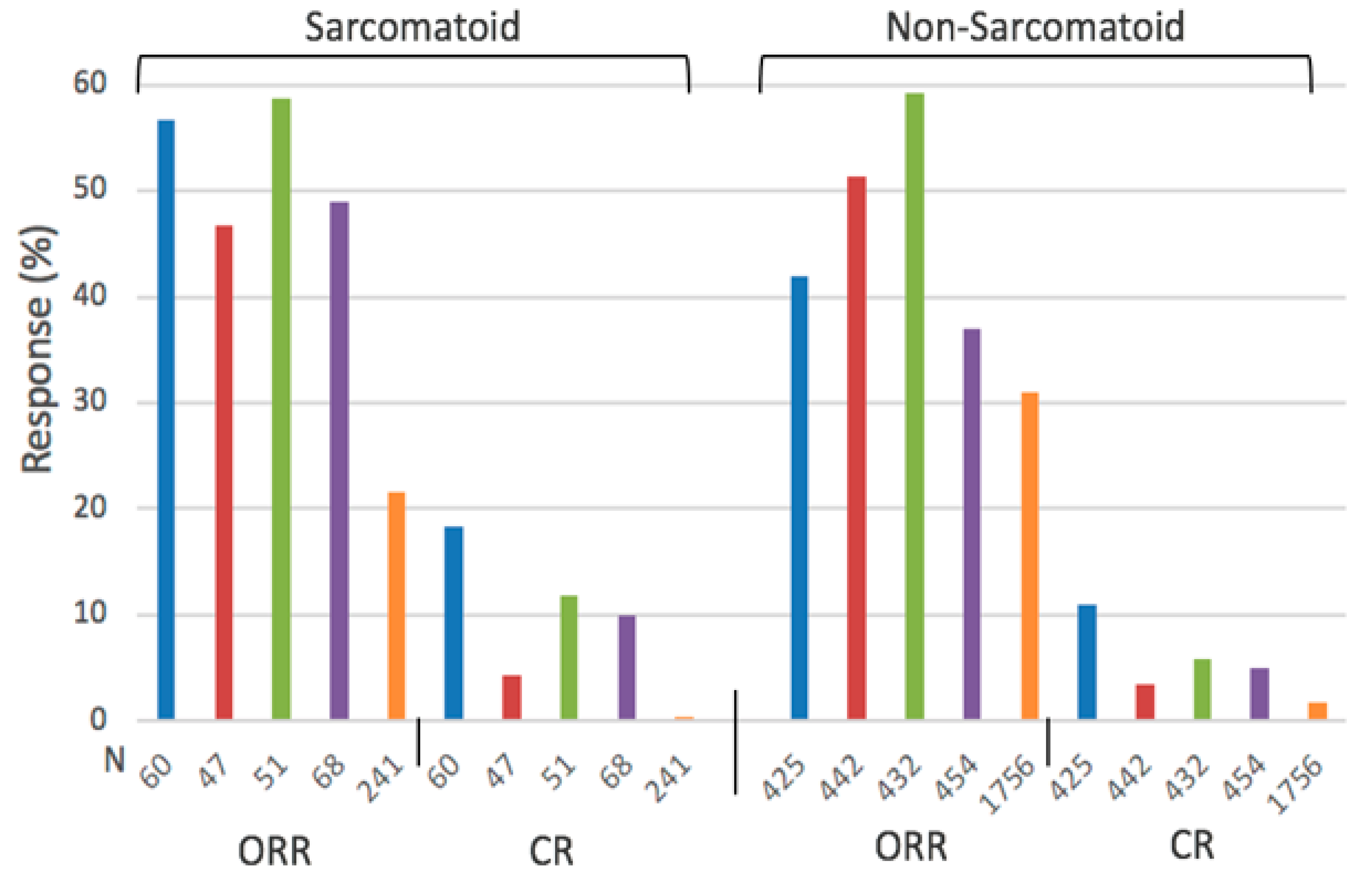

| Outcome | Patients with Sarc. Differentiation | Patients without Sarc. Differentiation | ||
|---|---|---|---|---|
| Combo | Sunitinib | Combo | Sunitinib | |
| ORR (%) | 52.8 (46.8–58.8) | 21.5 (14–31.5) | 47.6 (37–59.8) | 30.9 (25.7–35.7) |
| mPFS (Months) | 7.9 (7.0–8.4) | 5.7 (4.0–8.3) | 12.1 (8.2–15.1) | 9.1 (8.3–11.1) |
| mOS (Months) | 24.8 (18.3, 31.2) | 14.3 (13.6, 18.3) | 34.6 (33.6, 35.6) | 30.8 (26.6, 34.9) |
| Site | n | Positive Nrf2 Expression |
|---|---|---|
| ccRCC tumor | 152 | 119 (78.3%) |
| Normal tissue | 151 | 87 (57.6%) |
| Tissue | n | Median TP Activity (u/mg Protein) |
|---|---|---|
| RCC | 116 | 12.8 (3.2–933.9) |
| Kidney | 90 | 11.79 (0–128.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rustum, Y.M.; Reis, R.; Rustum, T.M. Druggable Biomarkers Altered in Clear Cell Renal Cell Carcinoma: Strategy for the Development of Mechanism-Based Combination Therapy. Int. J. Mol. Sci. 2023, 24, 902. https://doi.org/10.3390/ijms24020902
Rustum YM, Reis R, Rustum TM. Druggable Biomarkers Altered in Clear Cell Renal Cell Carcinoma: Strategy for the Development of Mechanism-Based Combination Therapy. International Journal of Molecular Sciences. 2023; 24(2):902. https://doi.org/10.3390/ijms24020902
Chicago/Turabian StyleRustum, Youcef M., Ryan Reis, and Tara M. Rustum. 2023. "Druggable Biomarkers Altered in Clear Cell Renal Cell Carcinoma: Strategy for the Development of Mechanism-Based Combination Therapy" International Journal of Molecular Sciences 24, no. 2: 902. https://doi.org/10.3390/ijms24020902