
Docking and Electronic Structure of Rutin, Myricetin, and Baicalein Targeting 3CLpro



Abstract
:1. Introduction
2. Results and Discussion
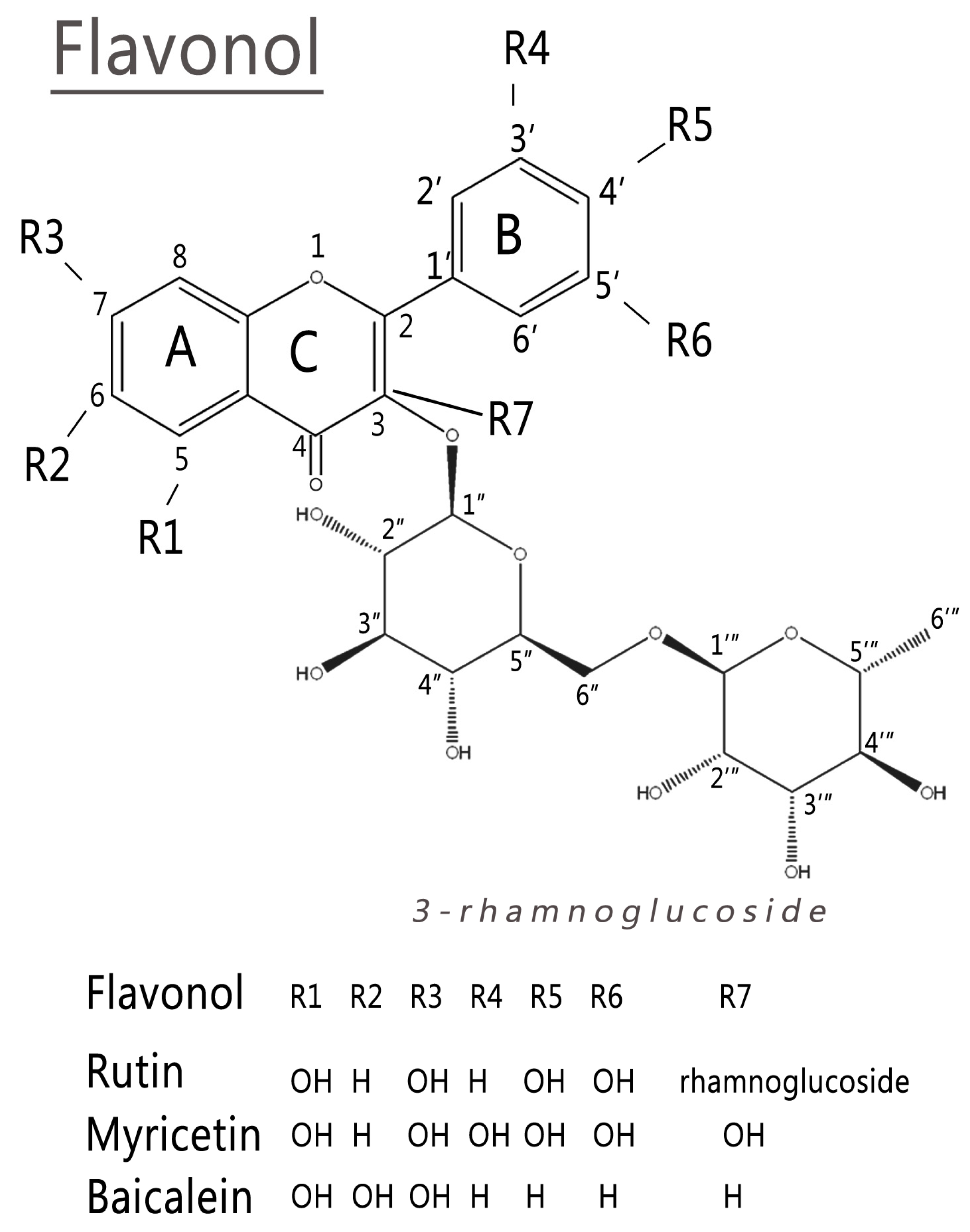
2.1. Conformational Analysis
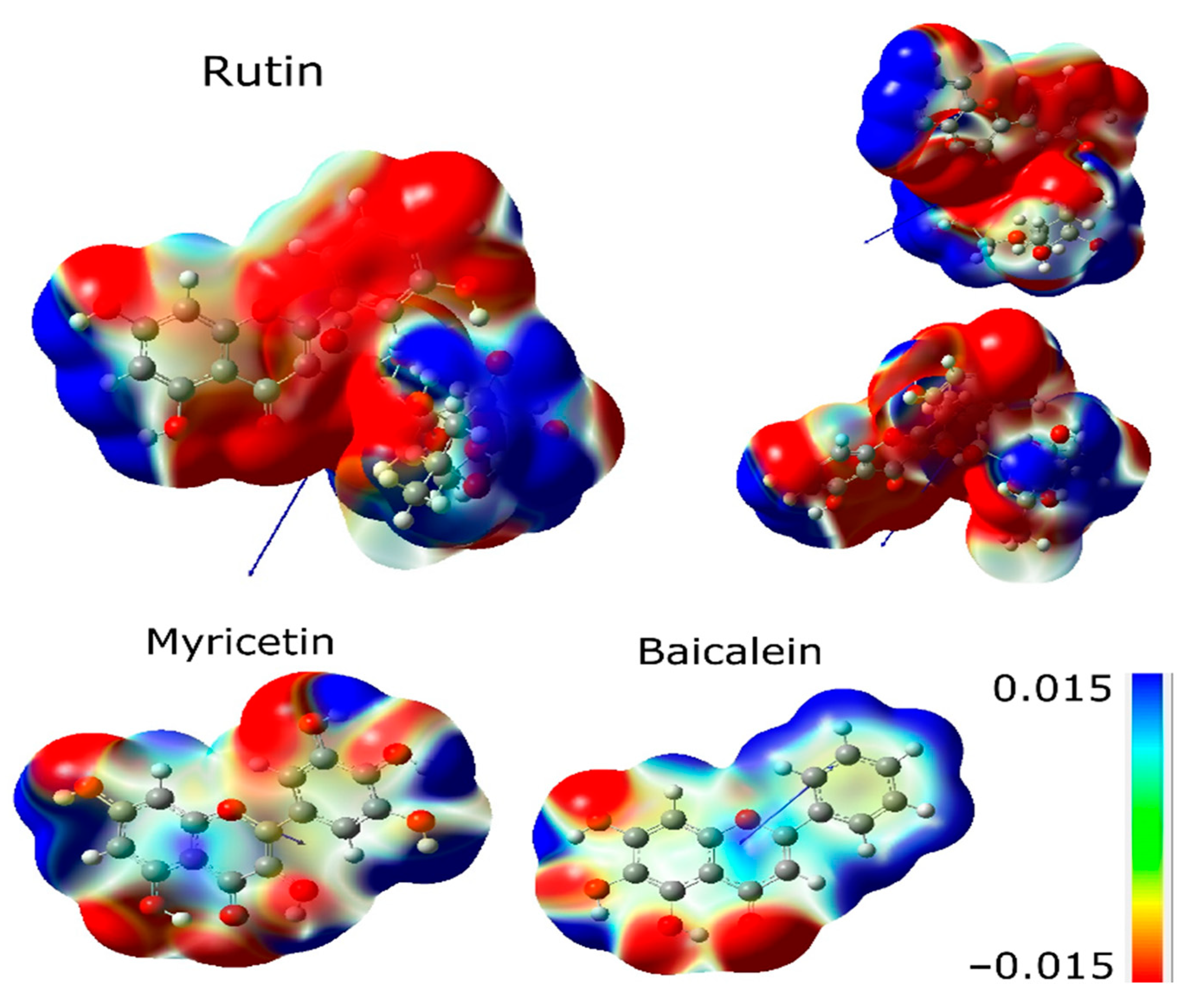
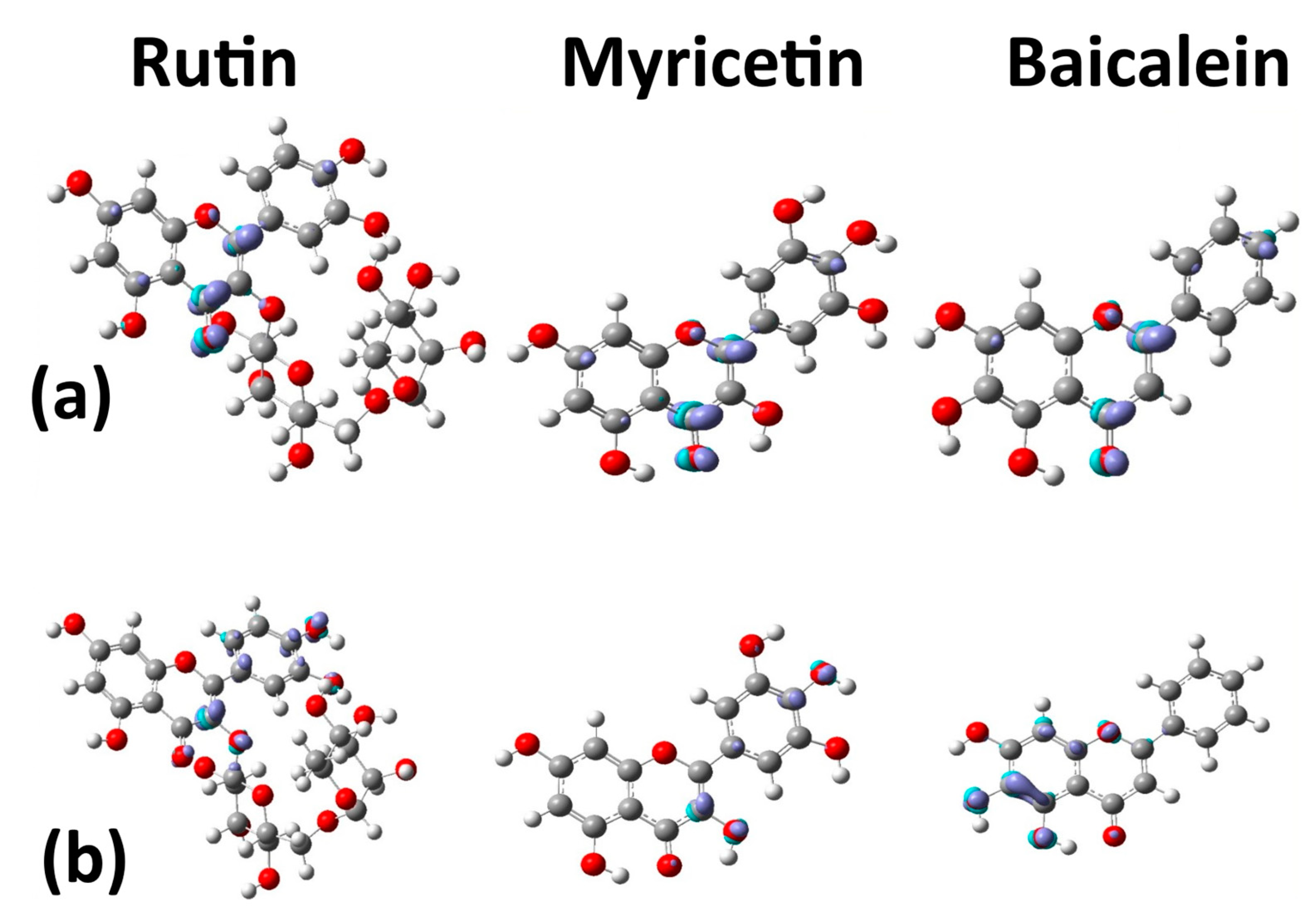
2.2. Electronic Analysis
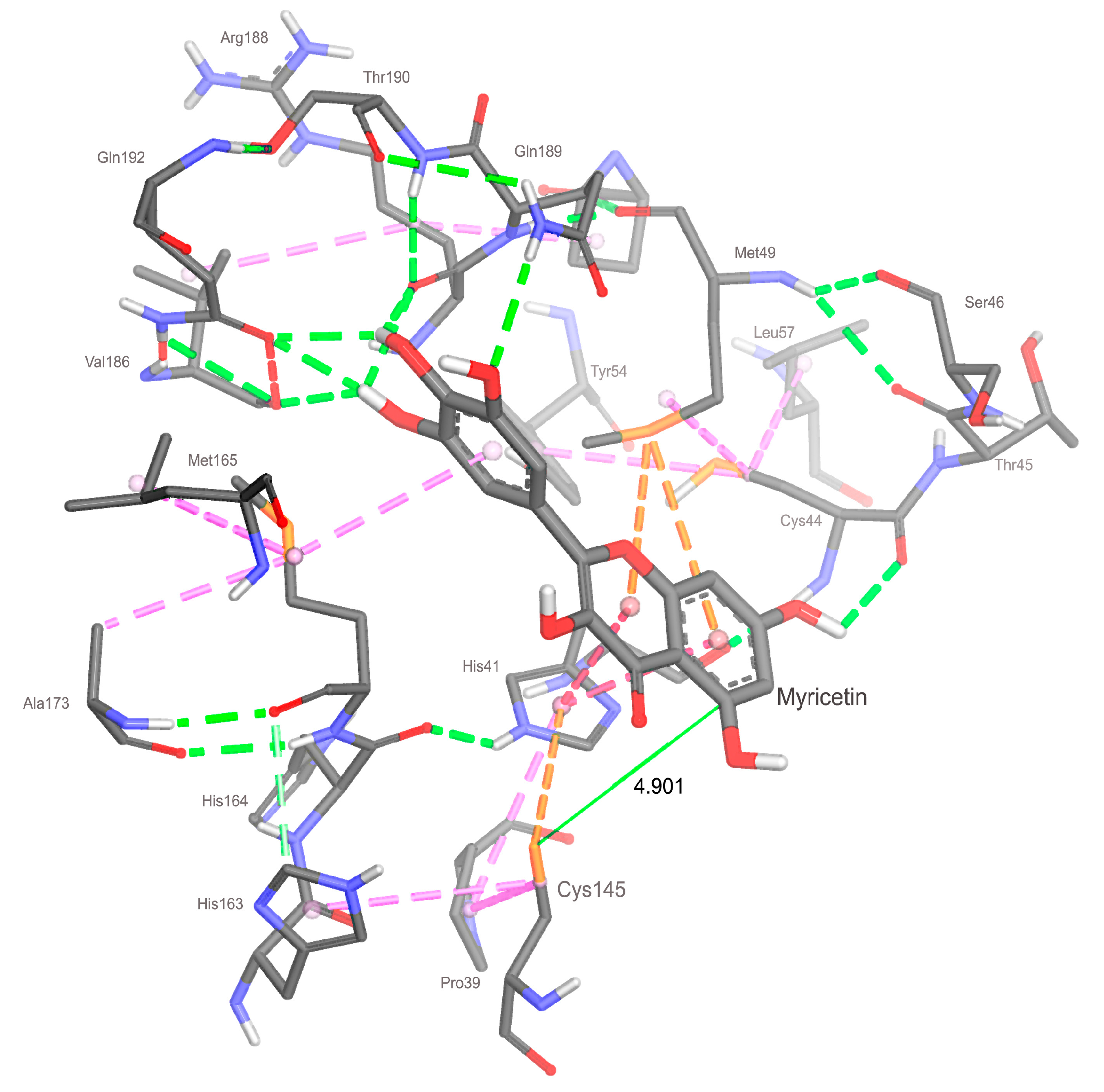
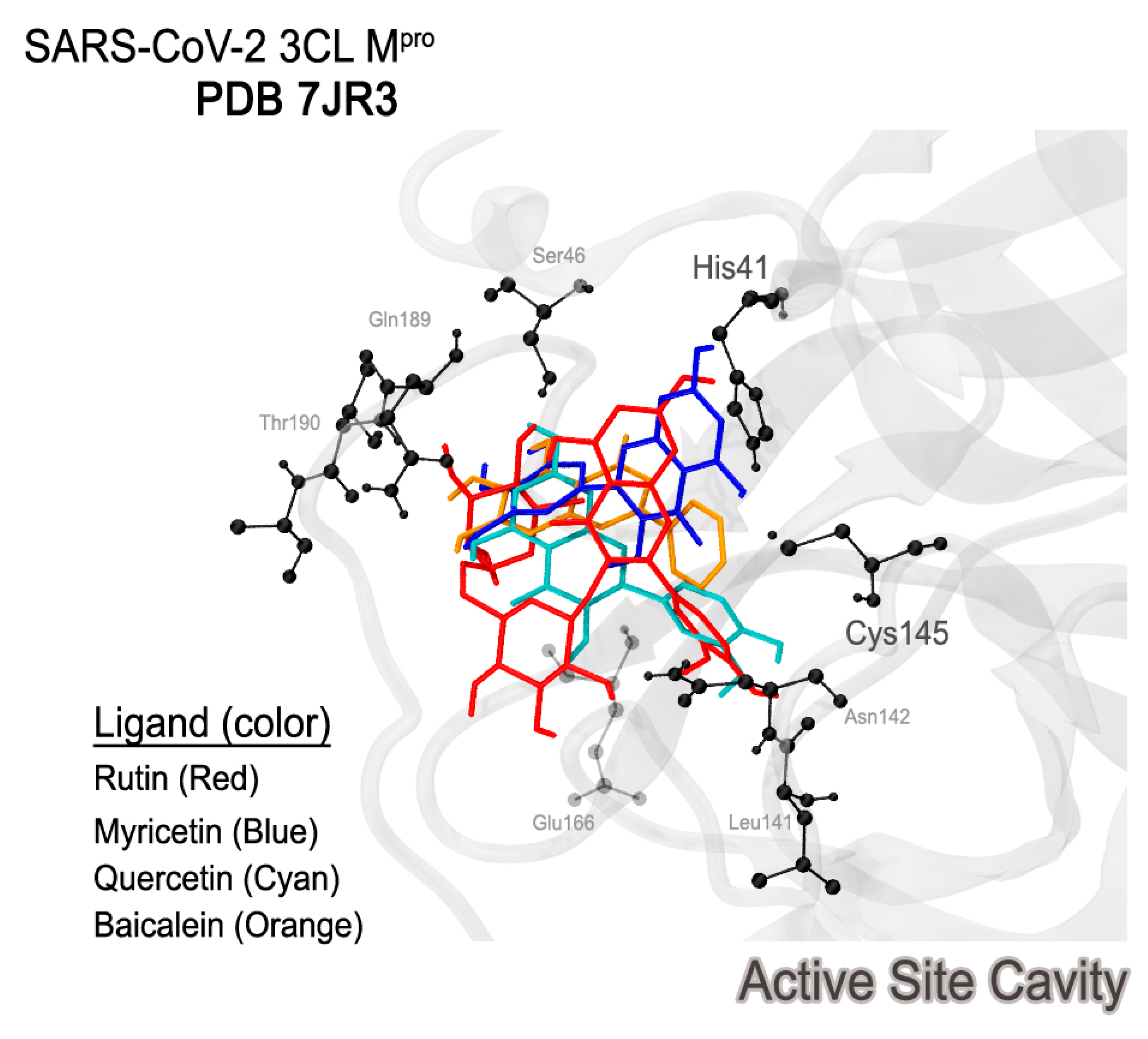
2.3. Docking
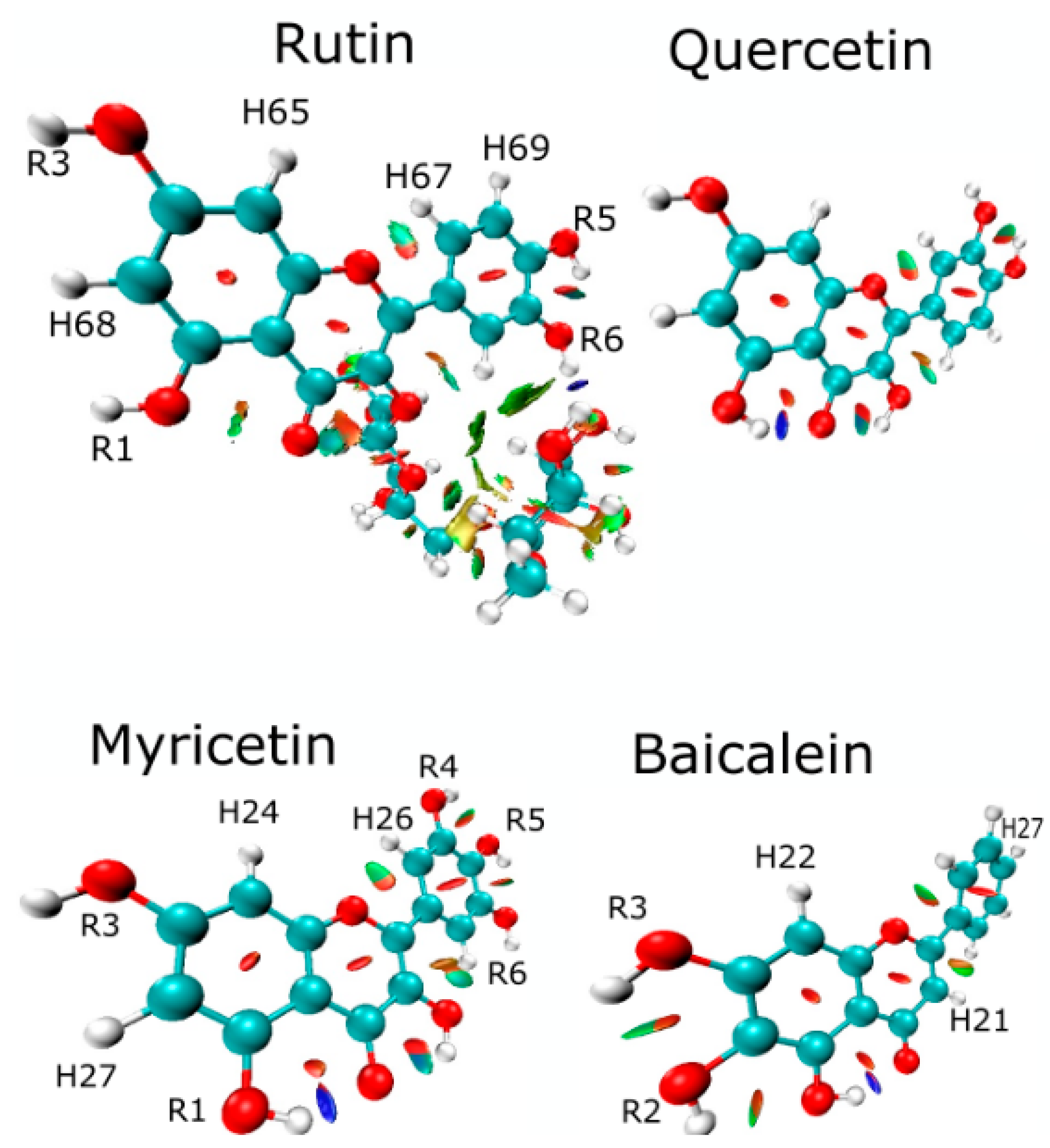
2.4. Analyzing Electronic Structure against Docking Information
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abian, O.; Ortega-Alarcon, D. Structural Stability of SARS-CoV-2 3CLpro and Identification of Quercetin as an Inhibitor by Experimental Screening. Int. J. Biol. Macromol. 2020, 164, 1693–1703. [Google Scholar]
- Rizzuti, B.; Grande, F.; Conforti, F.; Jimenez-Alesanco, A.; Ceballos-Laita, L.; Ortega-Alarcon, D.; Vega, S.; Reyburn, H.T.; Abian, O.; Velazquez-Campoy, A. Rutin Is a Low Micromolar Inhibitor of SARS-CoV-2 Main Protease 3CLpro: Implications for Drug Design of Quercetin Analogs. Biomedicines 2021, 9, 375. [Google Scholar]
- Deetanya, P.; Kowit Hengphasatporn, P.W.; Shigeta, Y.; Rungrotmongkol, T.; Wangkanont, K. Interaction of 8-Anilinonaphthalene-1-Sulfonate with SARS-CoV-2 Main Protease and Its Application as a Fluorescent Probe for Inhibitor Identification. Comput. Struct. Biotechnol. J. 2021, 19, 3364–3371. [Google Scholar]
- Gasmi, A.; Mujawdiya, P.K.; Lysiuk, R.; Shanaida, M.; Peana, M.; Gasmi Benahmed, A.; Beley, N.; Kovalska, N.; Bjørklund, G. Quercetin in the Prevention and Treatment of Coronavirus Infections: A Focus on SARS-CoV-2. Pharmaceuticals 2022, 15, 1049. [Google Scholar] [CrossRef]
- Bahun, M.; Jukić, M.; Oblak, D.; Kranjc, L.; Bajc, G.; Butala, M.; Bozovičar, K.; Bratkovič, T.; Podlipnik, Č.; Ulrih, N.P. Inhibition of the SARS-CoV-2 3CLpro Main Protease by Plant Polyphenols. Food Chem. 2022, 373, 131594. [Google Scholar] [CrossRef]
- Bhati, S.; Kaushik, V.; Singh, J. Rational Design of Flavonoid Based Potential Inhibitors Targeting SARS-CoV 3CL Protease for the Treatment of COVID-19. J. Mol. Struct. 2021, 1237, 130380. [Google Scholar] [CrossRef]
- Mori, M.; Quaglio, D.; Calcaterra, A.; Ghirga, F.; Sorrentino, L.; Cammarone, S.; Fracella, M.; D’Auria, A.; Frasca, F.; Criscuolo, E.; et al. Natural Flavonoid Derivatives Have Pan-Coronavirus Antiviral Activity. Microorganisms 2023, 11, 314. [Google Scholar] [CrossRef]
- Ghiasi, M.; Taheri, S.; Tafazzoli, M. Dynamic Stereochemistry of Rutin (Vitamin P) in Solution: Theoretical Approaches and Experimental Validation. Carbohydr. Res. 2010, 345, 1760–1766. [Google Scholar]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Jia Liu, Q.S.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; Ke, C.; et al. Identification of Pyrogallol as a Warhead in Design of Covalent Inhibitors for the SARS-CoV-2 3CL Protease. Nat. Commun. 2021, 12, 3623. [Google Scholar]
- Su, H.; Yao, S.; Zhao, W.; Li, M.; Liu, J.; Shang, W.S.; Xie, H.; Ke, C.; Hu, H.; Gao, M.; et al. Anti-SARS-CoV-2 Activities in Vitro of Shuanghuanglian Preparations and Bioactive Ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential Bioactive Glycosylated Flavonoids as SARS-CoV-2 Main Protease Inhibitors: A molecular Docking and Simulation Studies. PLoS ONE 2020, 15, e0240653. [Google Scholar] [CrossRef] [PubMed]
- da Silva, F.M.A.; da Silva, K.P.A.; de Oliveira, L.P.M.; Costa, E.V.; Koolen, H.H.F.; Pinheiro, M.L.B.; de Souza, A.Q.L.; de Souza, A.D.L. Flavonoid Glycosides and Their Putative Human Metabolites as Potential Inhibitors of the SARS-CoV-2 Main Protease (Mpro) and Rna-Dependent Rna Polymerase (Rdrp). Mem. Inst. Oswaldo Cruz 2020, 115, 115. [Google Scholar] [CrossRef] [PubMed]
- de Souza Farias, S.A.; da Costa, K.S.; Martins, J.B. Comparative Analysis of the Reactivity of Anthocyanidins, Leucoanthocyanidins, and Flavonols Using a Quantum Chemistry Approach. J. Mol. Model. 2023, 29, 93. [Google Scholar] [CrossRef]
- Ganeshpurkar, A.; Saluja, A.K. The Pharmacological Potential of Rutin. Saudi Pharm. J. 2017, 25, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; AlAjmi, M.F.; Hussain, A. Natural Compounds as Inhibitors of SARS-CoV-2 Main Protease (3CLpro): A Molecular Docking and Simulation Approach to Combat COVID-19. Curr. Pharm. Des. 2020, 27, 3577–3589. [Google Scholar] [CrossRef]
- Khalifa, I.; Zhu, W.; Mohammed, H.H.H.; Dutta, K.; Li, C. Tannins Inhibit SARS-CoV-2 through Binding with Catalytic Dyad Residues of 3CLpro: An In Silico Approach with 19 Structural Different Hydrolysable Tannins. J. Food Biochem. 2020, 44, e13432. [Google Scholar] [CrossRef]
- Khalifa, I.; Nawaz, A.; Sobhy, R.; Althawb, S.A.; Barakat, H. Polyacylated Anthocyanins Constructively Network with Catalytic Dyad Residues of 3CLpro of 2019-NCoV than Monomeric Anthocyanins: A Structural-Relationship Activity Study with 10 Anthocyanins Using in-Silico Approaches. J. Mol. Graph. Model. 2020, 100, 107690. [Google Scholar] [CrossRef]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria Baicalensis Extract and Baicalein Inhibit Replication of SARS-CoV-2 and Its 3C-like Protease in Vitro. J. Enzym. Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef]
- de Souza Farias, S.A.; Santane, K.C.; Martins, B.L.J. Analysis Conformational, Structural, Magnetic, and Eletronic Properties Related to Antioxidant Activity: Revisiting Flavan, Anthocyanidin, Flavone, Flavonol, Isoflavone and Flavan-3-Ol. ACS Omega 2021, 13, 8908–8918. [Google Scholar] [CrossRef]
- De Oliveira, R.R.; Nascimento, A.F.Z.; Zeri, A.C.M.; Trivella, D.B.B. SARS-CoV-2 3CL Protease Crystallized under Reducing Conditions. RCSB Protein Data Bank. 2020. Available online: https://pdbj.org/mine/summary/7jr3 (accessed on 25 August 2023).
- Michalík, M.; Biela, M.; Cagardová, D.; Lukeš, V. Influence of Catecholic Ring Torsion on Hydroxyflavones. Acta Chim. Slovaca 2020, 13, 49–55. [Google Scholar] [CrossRef]
- Cai, W.; Chen, Y.; Xie, L.; Zhang, H.; Hou, C. Characterization and Density Functional Theory Study of the Antioxidant Activity of Quercetin and Its Sugar-containing Analogues. Eur. Food Res. Technol. 2014, 238, 121–128. [Google Scholar] [CrossRef]
- Vojta, D.; Karlsen, E.M.; Spanget-Larsen, J. Electronic States of Myricetin. UV—Vis Polarization Spectroscopy and Quantum Chemical Calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 173, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Z.; Deng, G.; Guo, R.; Chen, D.F.; Wu, L.M. A DFT-Based Study of the Hydrogen-Bonding Interactions between Myricetin and Ethanol/Water. J. Mol. Model. 2019, 25, 67. [Google Scholar] [CrossRef] [PubMed]
- Sadasivam, K.; Kumaresan, R. Antioxidant Behavior of Mearnsetin and Myricetin Flavonoid Compounds—A DFT Study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 79, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Vojta, D.; Dominković, K.; Miljanić, S.; Spanget-Larsen, J. Intramolecular Hydrogen Bonding in Myricetin and Myricitrin. Quantum Chemical Calculations and Vibrational Spectroscopy. J. Mol. Struct. 2017, 1131, 242–249. [Google Scholar] [CrossRef]
- Justino, G.C.; Vieira, A.J.S.C. Antioxidant Mechanisms of Quercetin and Myricetin in the Gas Phase and in Solution- A Comparison and Validation of Semi-Empirical Methods. J. Mol. Model. 2010, 16, 863–876. [Google Scholar] [CrossRef]
- Freindorf, M.; Kraka, E.; Cremer, D. A Comprehensive Analysis of Hydrogen Bond Interactions Based on Local Vibrational Modes. Int. J. Quantum Chem. 2012, 112, 3174–3187. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density—Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chemie Int. Ed. English 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Martins, J.B.L.; Quintino, R.P.; José, R.; Politi, S.; Sethio, D.; Gargano, R. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy Computational Analysis of Vibrational Frequencies and Rovibrational Spectroscopic Constants of Hydrogen Sul Fi de Dimer Using MP2 and CCSD (T). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 239, 118540. [Google Scholar] [CrossRef]
- Matta, C.F.; Bader, R.F.W. Atoms-in-Molecules Study of the Genetically Encoded Amino Acids. III. Bond and Atomic Properties and Their Correlations with Experiment Including Mutation-Induced Changes in Protein Stability and Genetic Coding. Proteins Struct. Funct. Genet. 2003, 52, 360–399. [Google Scholar] [CrossRef]
- Wick, C.R.; Clark, T. On Bond-Critical Points in QTAIM and Weak Interactions. J. Mol. Model. 2018, 24, 142. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Sallum, L.O.; Menezes, A.C.S.; Camargo, A.J.; Napolitano, H.B. A Comprehensive Topological Analysis of a Novel Flavonoid Extracted from Brazilian Cerrado Plants. ChemistrySelect 2019, 4, 14012–14020. [Google Scholar] [CrossRef]
- Niquini, F.M.; Tenorio, J.C.; da Silva, M.F.G.F.; Ribeiro, A.B.; Wanderley, A.; Ellena, J.; Corrêa, R.S. On the Conformation, Molecular Interactions and Electron Density of a Natural Flavonoid Derivative. J. Mol. Struct. 2020, 1220, 128632. [Google Scholar] [CrossRef]
- Shirley, B.W. Flavonoid Biosynthesis: “New” Functions for an “Old” Pathway. Trends Plant Sci. 1996, 1, 377–382. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New HybridA New Hybrid Exchange—Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Matta, C.F.; Boyd, R.J. An Introduction to the Quantum Theory of Atoms in Molecules. In The Quantum Theory of Atoms in Molecules; Wiley Online Library: Hoboken, NJ, USA, 2007; pp. 1–34. ISBN 9783527610709. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Rizwana, B.F.; Muthu, S.; Prasana, J.C.; Abraham, C.S.; Raja, M. Spectroscopic (FT-IR, FT-Raman) Investigation, Topology (ESP, ELF, LOL) Analyses, Charge Transfer Excitation and Molecular Docking (Dengue, HCV) Studies on Ribavirin. Chem. Data Collect. 2018, 17–18, 236–250. [Google Scholar] [CrossRef]
- Jayasudha, J.; Balachandran, V.; Narayana, B. Molecular Docking, Spectroscopic, and Computational Studies of 2-{3-(4-Chlorophenyl)-5-[4-(propan-2-yl)phenyl]-4,5-dihydro-1H-pyrazol-1-yl}-1,3-thiazol-4(5H)-one. Polycycl. Aromat. Compd. 2020, 42, 2222–2244. [Google Scholar] [CrossRef]
- Srivastava, A.; Rawat, P.; Tandon, P.; Singh, R.N. A Computational Study on Conformational Geometries, Chemical Reactivity and Inhibitor Property of an Alkaloid Bicuculline with Gama-Aminobutyric Acid (GABA) by DFT. Comput. Theor. Chem. 2012, 993, 80–89. [Google Scholar] [CrossRef]
- Arulaabaranam, K.; Mani, G.; Muthu, S. Computational Assessment on Wave Function (ELF, LOL) Analysis, Molecular Confirmation and Molecular Docking Explores on 2-(5-Amino-2-methylanilino)-4-(3-pyridyl)pyrimidine. Chem. Data Collect. 2020, 29, 100525. [Google Scholar] [CrossRef]
- Prasana, J.C.; Muthu, S.; Abraham, C.S. Molecular Docking Studies, Charge Transfer Excitation and Wave Function Analyses (ESP, ELF, LOL) on Valacyclovir: A Potential Antiviral Drug. Comput. Biol. Chem. 2019, 78, 9–17. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, W.; Qi, L.; Ren, Y.; Ma, H. Investigation on 4-Amino-5-Substituent-1,2,4-Triazole-3-Thione Schiff Bases an Antifungal Drug by Characterization (Spectroscopic, XRD), Biological Activities, Molecular Docking Studies and Electrostatic Potential (ESP). J. Mol. Struct. 2019, 1197, 171–182. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardsona, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 1, 12–21. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- BIOVIA Dassault Systèmes, R. D. S. Discovery Studio Modeling Environment; Dassault Systèmes, R. D. S.: San Diego, CA, USA, 2017. [Google Scholar]
- Skoko, S.; Ambrosetti, M.; Giovannini, T.; Cappelli, C. Simulating Absorption Spectra of Flavonoids in Aqueous Solution: A Polarizable QM/MM Study. Molecules 2020, 25, 5853. [Google Scholar] [CrossRef]
- Aparicio, S. A Systematic Computational Study on Flavonoids. Int. J. Mol. Sci. 2010, 11, 2017–2038. [Google Scholar] [CrossRef]
- Zhang, H.; Zhai, J.; Zhang, L.; Li, C.; Zhao, Y.; Chen, Y.; Li, Q.; Hu, X.P. In Vitro Inhibition of Glyoxalase I by Flavonoids: New Insights from Crystallographic Analysis. Medlin. Curr. Top. Med. Chem. 1016, 16, 460–466. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S | |||||||
|---|---|---|---|---|---|---|---|
| Rutin | −159.74 | −15.54 | 144.20 | 87.64 | 72.10 | 0.001 | 53.27 |
| Myricetin | −172.52 | −33.82 | 138.70 | 103.16 | 69.35 | 0.001 | 76.73 |
| Myricetin * | −171.51 | −24.64 | 146.87 | 98.08 | 73.43 | 0.001 | 65.50 |
| Baicalein | −179.15 | −30.03 | 149.12 | 104.58 | 74.56 | 0.001 | 73.35 |
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Rutin | 3.12 (0.24) | 2.55 (0.13) | 2.88 (−0.15) | 3.57 (0.17) |
| Myricetin | −4.88 (−0.11) | 2.96 (0.13) | 3.31 (0.17) | - |
| Myricetin * | 3.39 (0.22) | 2.95 (0.13) | - | - |
| Baicalein | −6.86 (−1.47) | - | - | - |
| Score (kcal/mol) | MW (g/mol) | Rotatable Bonds | Haceptor | Hdonor | Log p | |
|---|---|---|---|---|---|---|
| Baicalein | −7.0 | 273.26 | 1 | 5 | 3 | 0.28 |
| Quercetin | −7.1 | 302.24 | 1 | 7 | 5 | 1.23 |
| Myricetin | −7.4 | 319.24 | 1 | 8 | 6 | 0.27 |
| Rutin | −8.6 | 610.52 | 6 | 16 | 10 | −1.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farias, S.A.d.S.; Rocha, K.M.L.; Nascimento, É.C.M.; de Jesus, R.d.C.C.; Neres, P.R.; Martins, J.B.L. Docking and Electronic Structure of Rutin, Myricetin, and Baicalein Targeting 3CLpro. Int. J. Mol. Sci. 2023, 24, 15113. https://doi.org/10.3390/ijms242015113
Farias SAdS, Rocha KML, Nascimento ÉCM, de Jesus RdCC, Neres PR, Martins JBL. Docking and Electronic Structure of Rutin, Myricetin, and Baicalein Targeting 3CLpro. International Journal of Molecular Sciences. 2023; 24(20):15113. https://doi.org/10.3390/ijms242015113
Chicago/Turabian StyleFarias, Sergio A. de S., Kelvyn M. L. Rocha, Érica C. M. Nascimento, Rafael do C. C. de Jesus, Paulo R. Neres, and João B. L. Martins. 2023. "Docking and Electronic Structure of Rutin, Myricetin, and Baicalein Targeting 3CLpro" International Journal of Molecular Sciences 24, no. 20: 15113. https://doi.org/10.3390/ijms242015113
APA StyleFarias, S. A. d. S., Rocha, K. M. L., Nascimento, É. C. M., de Jesus, R. d. C. C., Neres, P. R., & Martins, J. B. L. (2023). Docking and Electronic Structure of Rutin, Myricetin, and Baicalein Targeting 3CLpro. International Journal of Molecular Sciences, 24(20), 15113. https://doi.org/10.3390/ijms242015113