
Genomes of a Novel Group of Phages That Use Alternative Genetic Code Found in Human Gut Viromes


 , , ,
, , ,
Abstract
:1. Introduction
2. Results
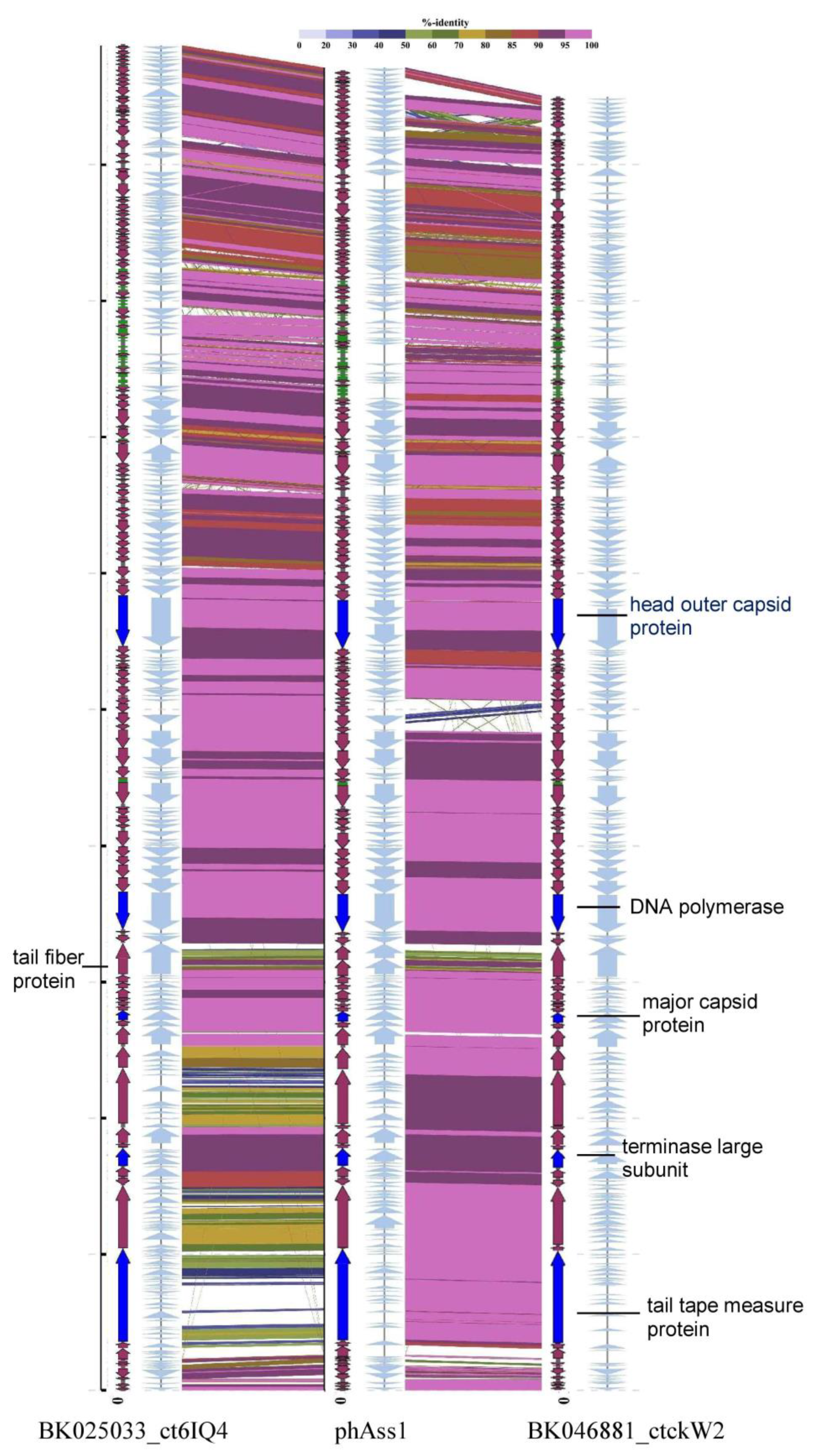
2.1. Analysis of the phAss-1 Bacteriophage Genome
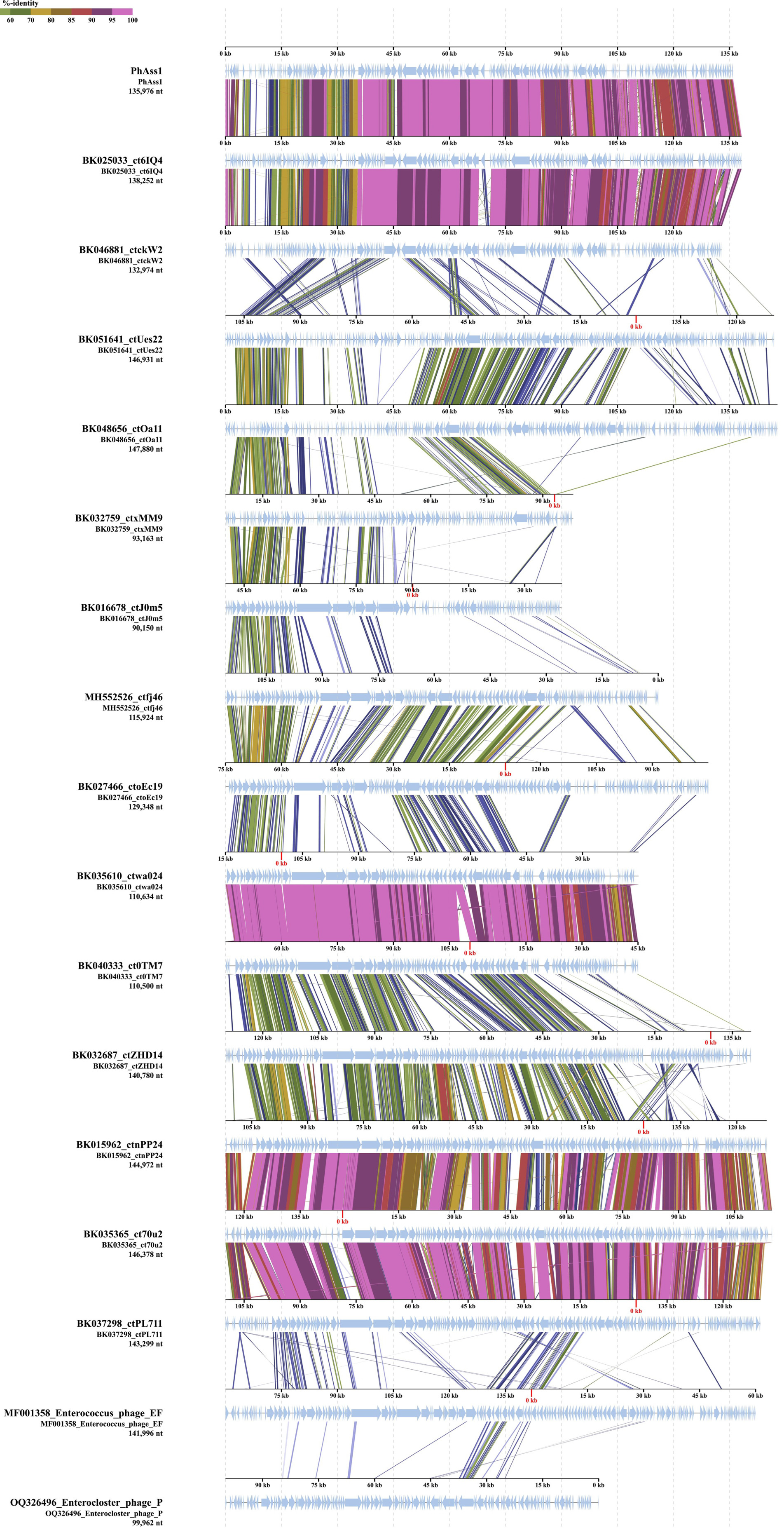
2.2. phAss-1 Virome Comparative Analysis
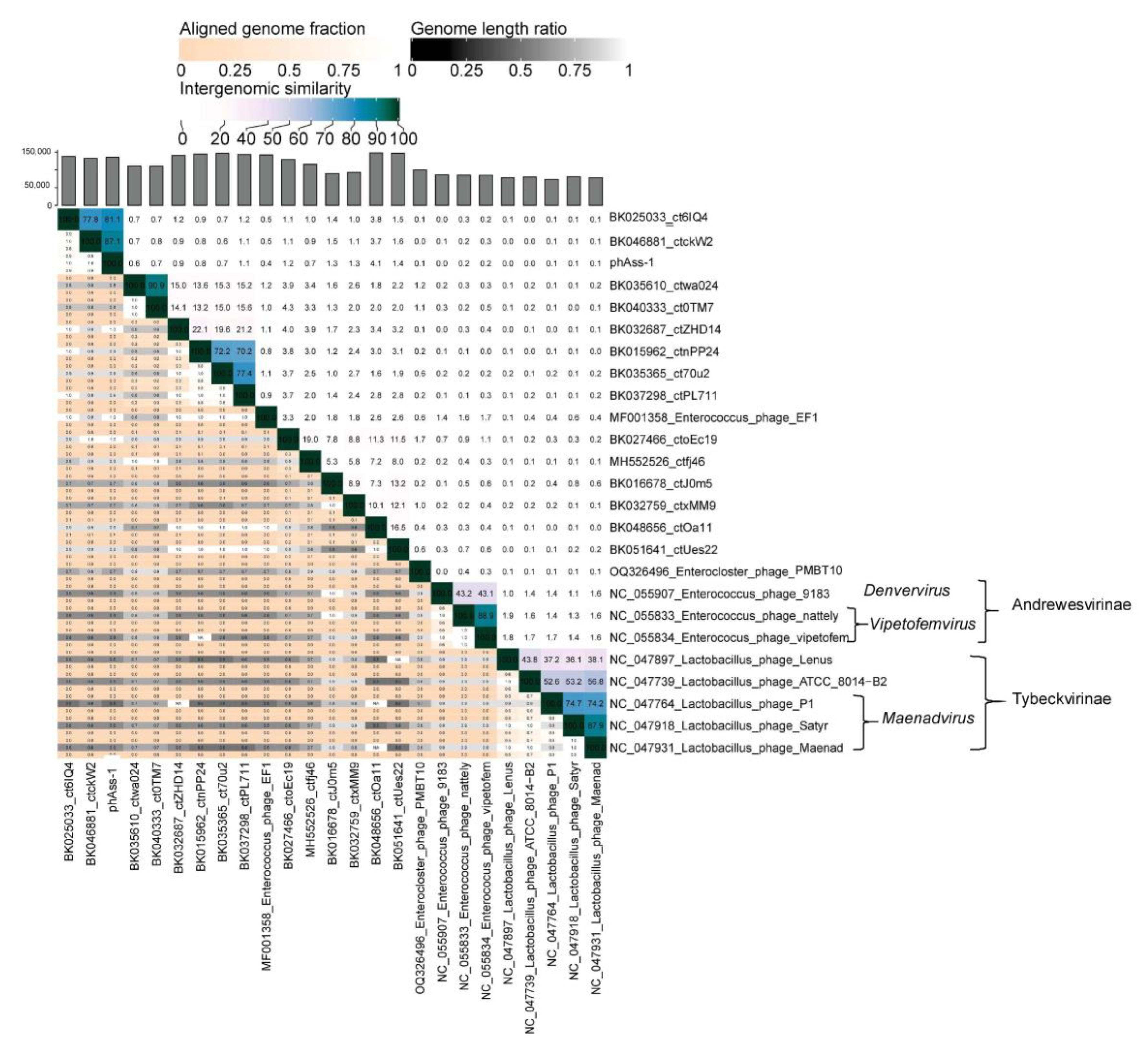
2.3. VIRIDIC Clustering
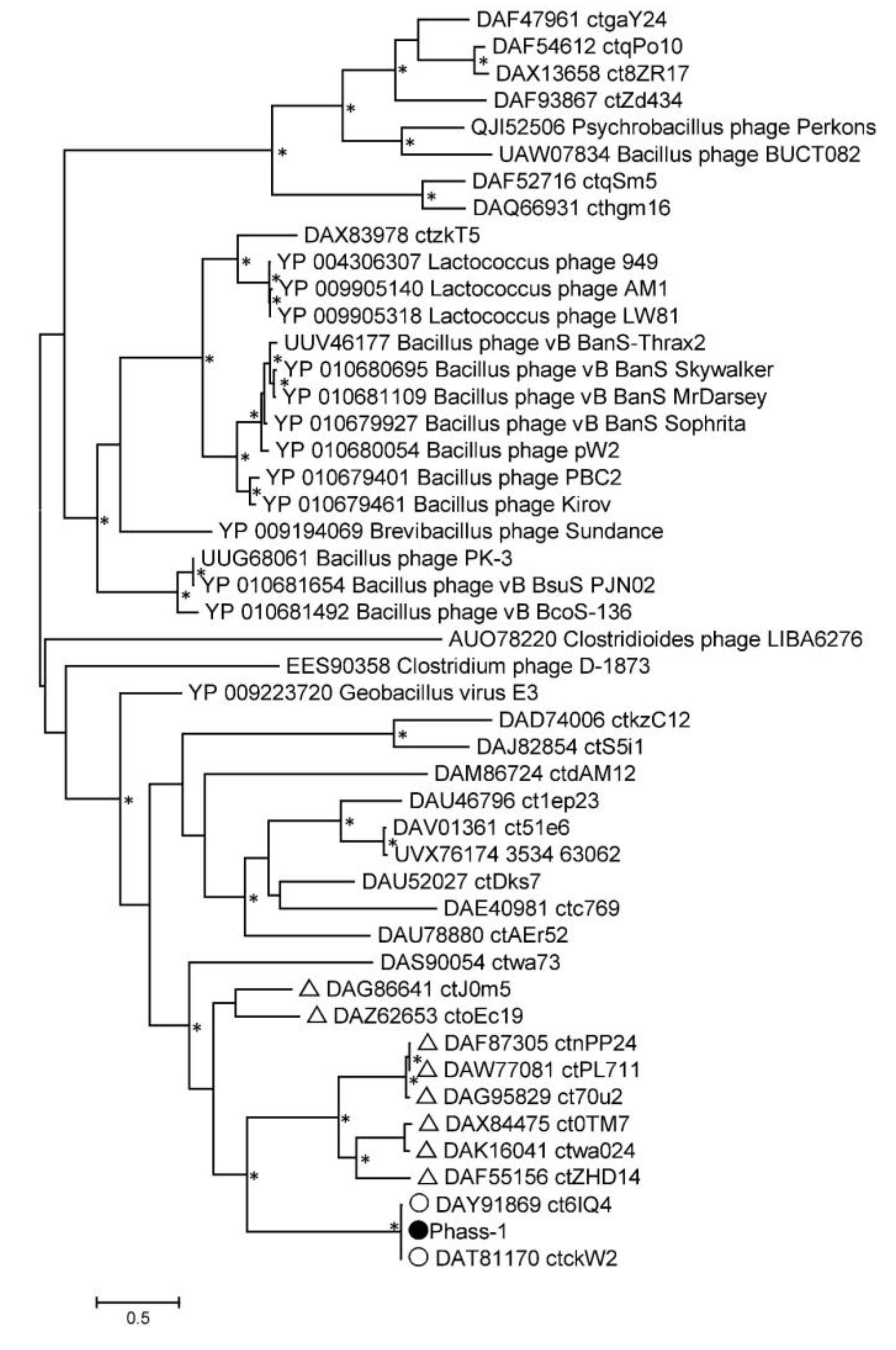
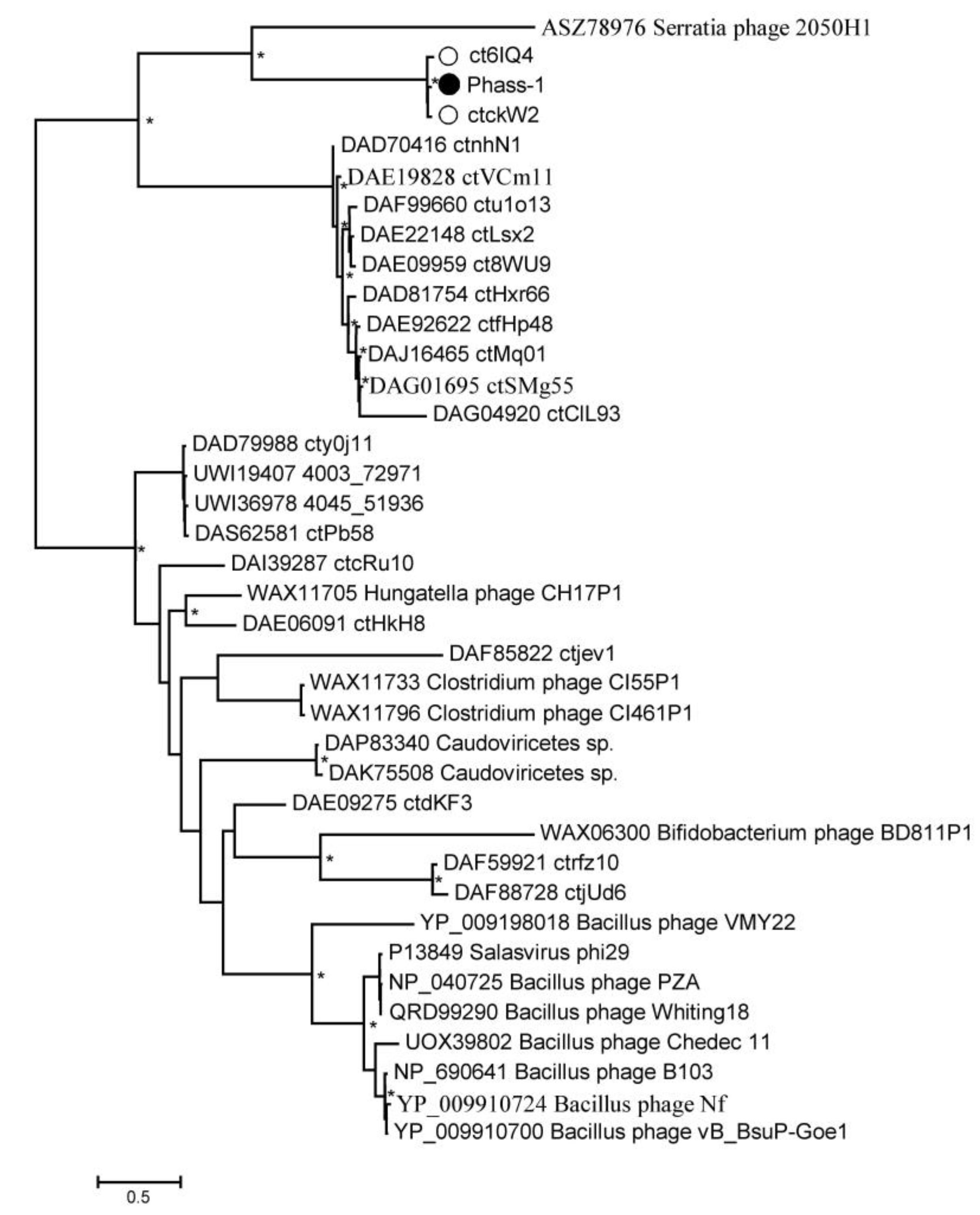
2.4. Phylogenetic Analysis of the phAss-1 Proteins
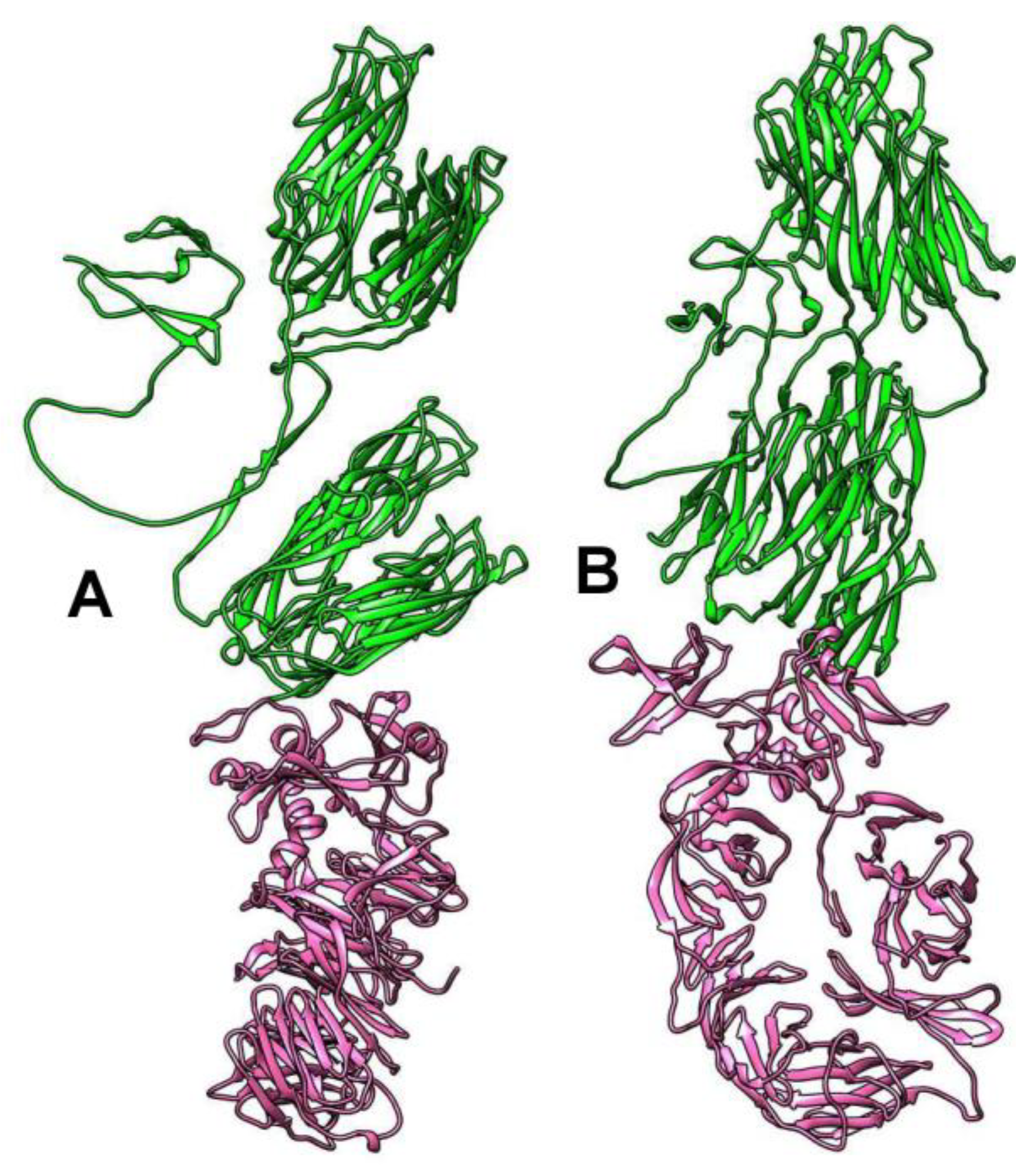
2.5. In Silico Host Prediction
3. Discussion
4. Materials and Methods
4.1. Virome Sequencing
4.2. Genome Analysis
4.3. Comparative Analysis
4.4. Phylogenetic Analysis
4.5. 3D Modeling of Protein Structure
4.6. In silico Host Prediction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. 2020, e00061-19. [Google Scholar] [CrossRef]
- Townsend, E.M.; Kelly, L.; Muscatt, G.; Box, J.D.; Hargraves, N.; Lilley, D.; Jameson, E. The Human Gut Phageome: Origins and Roles in the Human Gut Microbiome. Front. Cell Infect. Microbiol. 2021, 4, 643214. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, C.B.; Shkoporov, A.N.; Upadrasta, A.; Khokhlova, E.V.; Ross, R.P.; Hill, C. Probing the “Dark Matter” of the Human Gut Phageome: Culture Assisted Metagenomics Enables Rapid Discovery and Host-Linking for Novel Bacteriophages. Front. Cell Infect. Microbiol. 2021, 11, 616918. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef]
- Yutin, N.; Benler, S.; Shmakov, S.A.; Wolf, Y.I.; Tolstoy, I.; Rayko, M.; Antipov, D.; Pevzner, P.A.; Koonin, E.V. Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features. Nat. Commun. 2021, 12, 1044. [Google Scholar] [CrossRef]
- Devoto, A.E.; Santini, J.M.; Olm, M.R.; Anantharaman, K.; Munk, P.; Tung, J.; Archie, E.A.; Turnbaugh, P.J.; Seed, K.D.; Blekhman, B.; et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 2019, 4, 693–700. [Google Scholar] [CrossRef]
- Borges, A.L.; Lou, Y.C.; Sachdeva, R.; Al-Shayeb, B.; Penev, P.I.; Jaffe, A.L.; Lei, S.; Santini, J.M.; Banfield, J.F. Widespread stop-codon recoding in bacteriophages may regulate translation of lytic genes. Nat Microbiol. 2022, 7, 918–927. [Google Scholar] [CrossRef]
- Ivanova, N.N.; Schwientek, P.; Tripp, H.J.; Rinke, C.; Pati, A.; Huntemann, M.; Visel, A.; Woyke, T.; Kyrpides, N.C.; Rubin, E.M. Stop codon reassignments in the wild. Science 2014, 344, 909–913. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across Earth’s ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef]
- Crisci, M.A.; Chen, L.X.; Devoto, A.E.; Borges, A.L.; Bordin, N.; Sachdeva, R.; Tett, A.; Sharrar, A.M.; Segata, N.; Debenedetti, F.; et al. Closely related Lak megaphages replicate in the microbiomes of diverse animals. iScience 2021, 24, 102875. [Google Scholar] [CrossRef]
- Peters, S.L.; Borges, A.L.; Giannone, R.J.; Morowitz, M.J.; Banfield, J.F.; Hettich, R.L. Experimental validation that human microbiome phages use alternative genetic coding. Nat. Commun. 2022, 13, 5710. [Google Scholar] [CrossRef]
- Nayfach, S.; Páez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.C.; Castro-Nallar, E.; Fisher, J.N.; Breakwell, D.P.; Grose, J.H.; Burnett, S.H. Phage cluster relationships identified 595 through single gene analysis. BMC Genom. 2013, 14, 410. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Search and Contextual Analysis of Transfer RNA Genes. Nucl. Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Bhunchoth, A.; Blanc-Mathieu, R.; Mihara, T.; Nishimura, Y.; Askora, A.; Phironrit, N.; Leksomboon, C.; Chatchawankanphanich, O.; Kawasaki, T.; Nakano, M.; et al. Two asian jumbo phages, ϕRSL2 and ϕRSF1, infect Ralstonia solanacearum and show common features of ϕKZ-related phages. Virology 2016, 494, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Tisza, M.J.; Buck, C.B. A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Šiborová, M.; Füzik, T.; Procházková, M.; Nováček, J.; Benešík, M.; Nilsson, A.S.; Plevka, P. Tail proteins of phage SU10 reorganize into the nozzle for genome delivery. Nat. Commun. 2022, 13, 5622. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Fàbrega-Ferrer, M.; Machón, C.; Conesa, J.J.; Fernández, F.J.; Pérez-Luque, R.; Pérez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 bacteriophage portal and tail suggest a viral DNA retention and ejection mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef]
- Scholl, D.; Merril, C. The genome of bacteriophage K1F, a T7-like phage that has acquired the ability to replicate on K1 strains of Escherichia coli. J. Bacteriol. 2005, 187, 8499–8503. [Google Scholar] [CrossRef] [PubMed]
- Morozova, V.; Kozlova, Y.; Shedko, E.; Kurilshikov, A.; Babkin, I.; Tupikin, A.; Yunusova, A.; Chernonosov, A.; Baykov, I.; Кondratov, I.; et al. Lytic bacteriophage PM16 specific for Proteus mirabilis: A novel member of the genus Phikmvvirus. Arch. Virol. 2016, 161, 2457–2472. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Brister, J.R. How to Name and Classify Your Phage: An Informal Guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. Viptree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | NCBI Accession Number and Name of Phage/Isolate | Genome Length | Source/Host | GC Content, % | SG | Mean Identity, % | Length, % |
|---|---|---|---|---|---|---|---|
| 1 | OQ749652_phAss-1 | 135,976 | human gut metagenome | 44.52 | 1 | 100 | 99.8 |
| 2 | BK046881_ctckW2 | 132,974 | human gut metagenome | 44.39 | 0.8612 | 92.7 | 92.3 |
| 3 | BK025033_ct6IQ4 | 138,252 | human gut metagenome | 44.45 | 0.8112 | 89.0 | 92.4 |
| 4 | BK048656_ctOa11 | 147,880 | human gut metagenome | 35.33 | 0.0975 | 46.4 | 20.8 |
| 5 | BK032759_ctxMM9 | 93,163 | human gut metagenome | 32.75 | 0.0741 | 43.4 | 11.2 |
| 6 | BK051641_ctUes22 | 146,931 | human gut metagenome | 33.03 | 0.0651 | 43.1 | 14.8 |
| 7 | MH552526_ctfj46 | 115,924 | mouse tissue metagenome | 30.75 | 0.0626 | 41.1 | 12 |
| 8 | BK027466_ctoEc19 | 129,348 | human gut metagenome | 28.95 | 0.0596 | 42.6 | 12.4 |
| 9 | BK016678_ctJ0m5 | 90,150 | human gut metagenome | 33.42 | 0.0528 | 42.3 | 7.8 |
| 10 | BK040333_ct0TM7 | 110,500 | human oral metagenome | 35.22 | 0.0507 | 40.5 | 9.5 |
| 11 | BK032687_ctZHD14 | 140,780 | human gut metagenome | 37.28 | 0.0502 | 42.8 | 11.5 |
| 12 | BK037298_ctPL711 | 143,299 | human gut metagenome | 38.20 | 0.0499 | 41.0 | 11.9 |
| 13 | BK035610_ctwa024 | 110,634 | human oral metagenome | 35.84 | 0.0496 | 40.8 | 9.4 |
| 14 | BK015962_ctnPP24 | 144,972 | human gut metagenome | 38.72 | 0.0466 | 40.8 | 11.4 |
| 15 | BK035365_ct70u2 | 146,378 | human gut metagenome | 38.36 | 0.0458 | 40.2 | 11.1 |
| 16 | MF001358_Enterococcus_ phage_EF | 141,996 | Enterococcus faecalis | 31.94 | 0.0281 | 39.1 | 6.9 |
| 17 | OQ326496_Enterocloster_ phage_P | 99,962 | Enterocloster bolteae Q14 | 32.11 | 0.0123 | 39.7 | 2.1 |
| № | Function Assigned to the Specific ORF * | Phage Genomes | ||
|---|---|---|---|---|
| phAss-1 | ctckW2 | ct6IQ4 | ||
| TAG suppressor stop codons | ||||
| 1 | ATP-dependent Clp protease | • | • | |
| 2 | Ribonucleoside-triphosphate reductase activating protein | • | ||
| 3 | Multiple antibiotic resistance protein MarR/DNA | • | ||
| 4 | PhoH family ribonuclease | • | • | |
| 5 | Phage holin | • | • | |
| 6 | L-shaped tail fiber protein | • | ||
| 7 | dUTPase | • | • | |
| 8 | NADAR family protein | • | • | |
| 9 | Antitermination protein, Q-dependent. | • | • | |
| 10 | P-loop containing nucleoside triphosphate hydrolase | • | n.d. ** | |
| 11 | Exoribonuclease | n.d. | • | |
| Presence in the genome | ||||
| 1 | Receptor-binding tail fiber protein (1070 aa) | • | • | |
| 2 | Structural tail protein (538 aa) | • | ||
| 3 | Receptor-binding tail fiber protein/pectatliase (429 aa) | • | ||
| 4 | LAGLIDADG endonuclease | • | ||
| 5 | Nicotinamidase | • | • | |
| 6 | Cysteine hydrolase | • | ||
| 7 | Transcription factor | • | ||
| 8 | Nucleotidyltransferase-like protein | • | • | |
| 9 | P-loop containing nucleoside triphosphate hydrolase | • | • | |
| 10 | 3′-5′ exonuclease | • | ||
| 11 | Exoribonuclease | • | • | |
| 12 | Phosphatase | • | • | |
| 13 | RNAse | • | • | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babkin, I.; Tikunov, A.; Morozova, V.; Matveev, A.; Morozov, V.V.; Tikunova, N. Genomes of a Novel Group of Phages That Use Alternative Genetic Code Found in Human Gut Viromes. Int. J. Mol. Sci. 2023, 24, 15302. https://doi.org/10.3390/ijms242015302
Babkin I, Tikunov A, Morozova V, Matveev A, Morozov VV, Tikunova N. Genomes of a Novel Group of Phages That Use Alternative Genetic Code Found in Human Gut Viromes. International Journal of Molecular Sciences. 2023; 24(20):15302. https://doi.org/10.3390/ijms242015302
Chicago/Turabian StyleBabkin, Igor, Artem Tikunov, Vera Morozova, Andrey Matveev, Vitaliy V. Morozov, and Nina Tikunova. 2023. "Genomes of a Novel Group of Phages That Use Alternative Genetic Code Found in Human Gut Viromes" International Journal of Molecular Sciences 24, no. 20: 15302. https://doi.org/10.3390/ijms242015302
APA StyleBabkin, I., Tikunov, A., Morozova, V., Matveev, A., Morozov, V. V., & Tikunova, N. (2023). Genomes of a Novel Group of Phages That Use Alternative Genetic Code Found in Human Gut Viromes. International Journal of Molecular Sciences, 24(20), 15302. https://doi.org/10.3390/ijms242015302