
Comprehensive Profiling of Alternative Splicing and Alternative Polyadenylation during Fruit Ripening in Watermelon (Citrullus lanatus)


 , ,
, ,
Abstract
:1. Introduction
2. Results
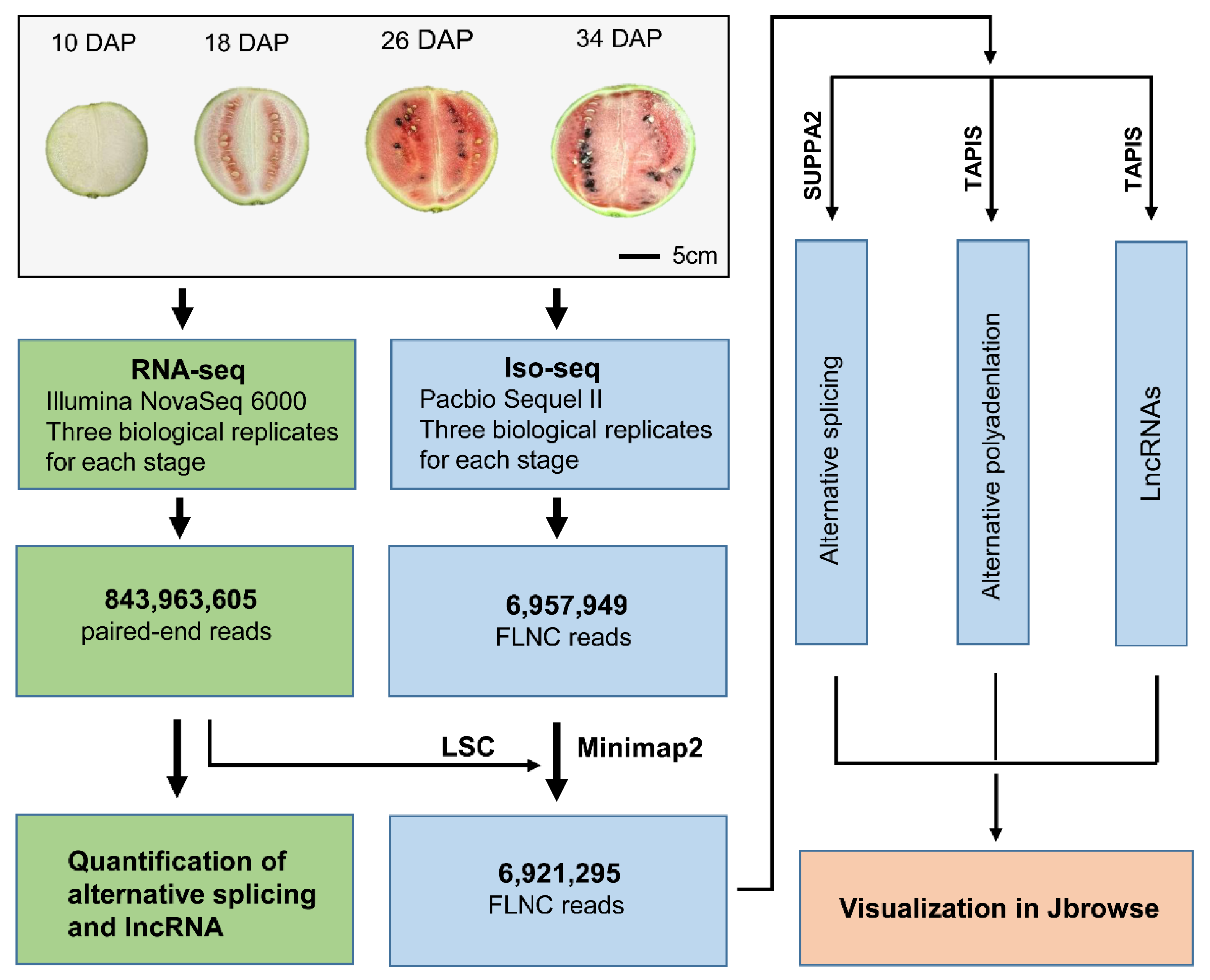
2.1. Watermelon Fruit Transcriptome Sequencing Using PacBio Iso-Seq
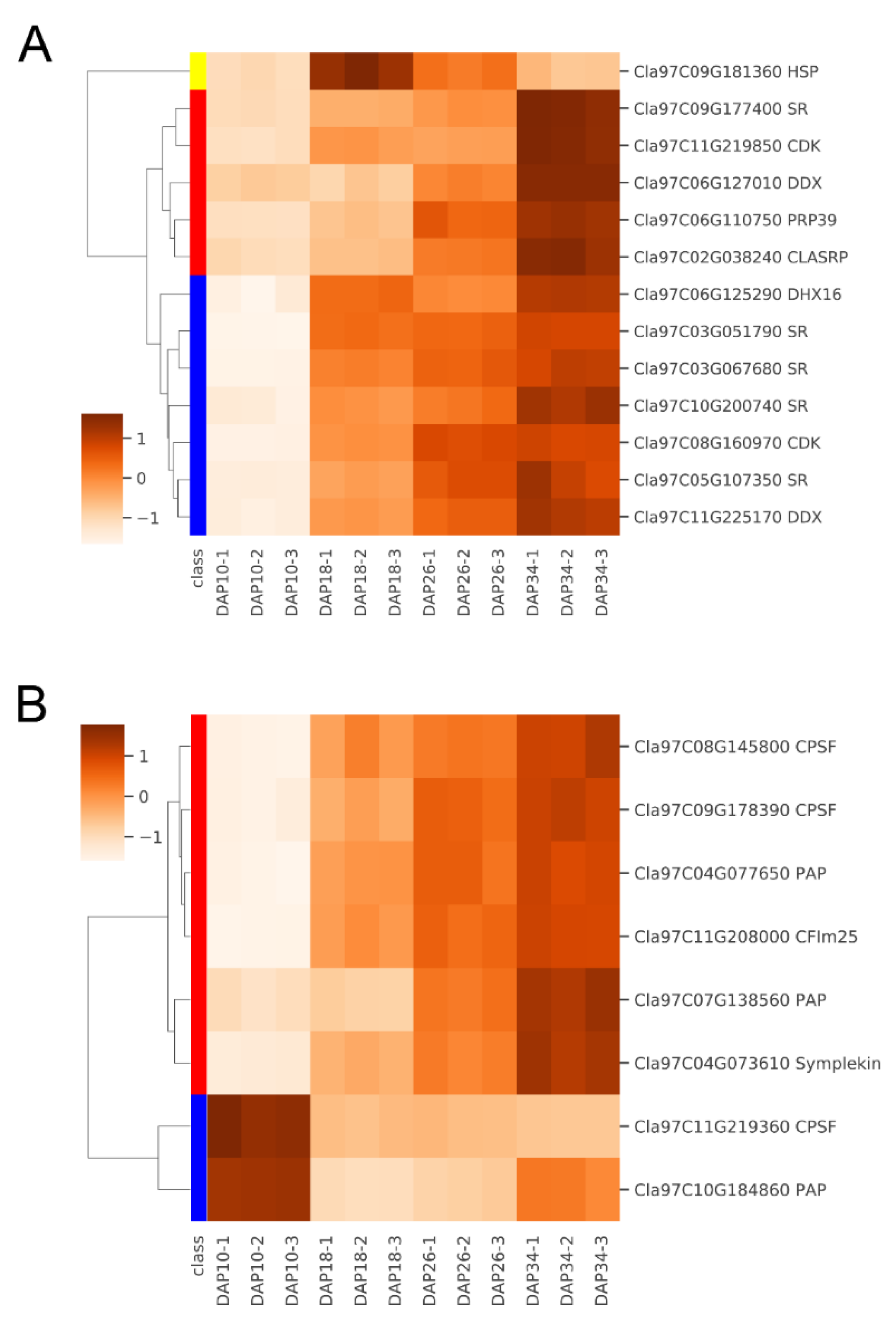
2.2. Identification of Full-Length Splicing Isoforms
2.3. Profiling of Global APA Sites and Differential APA
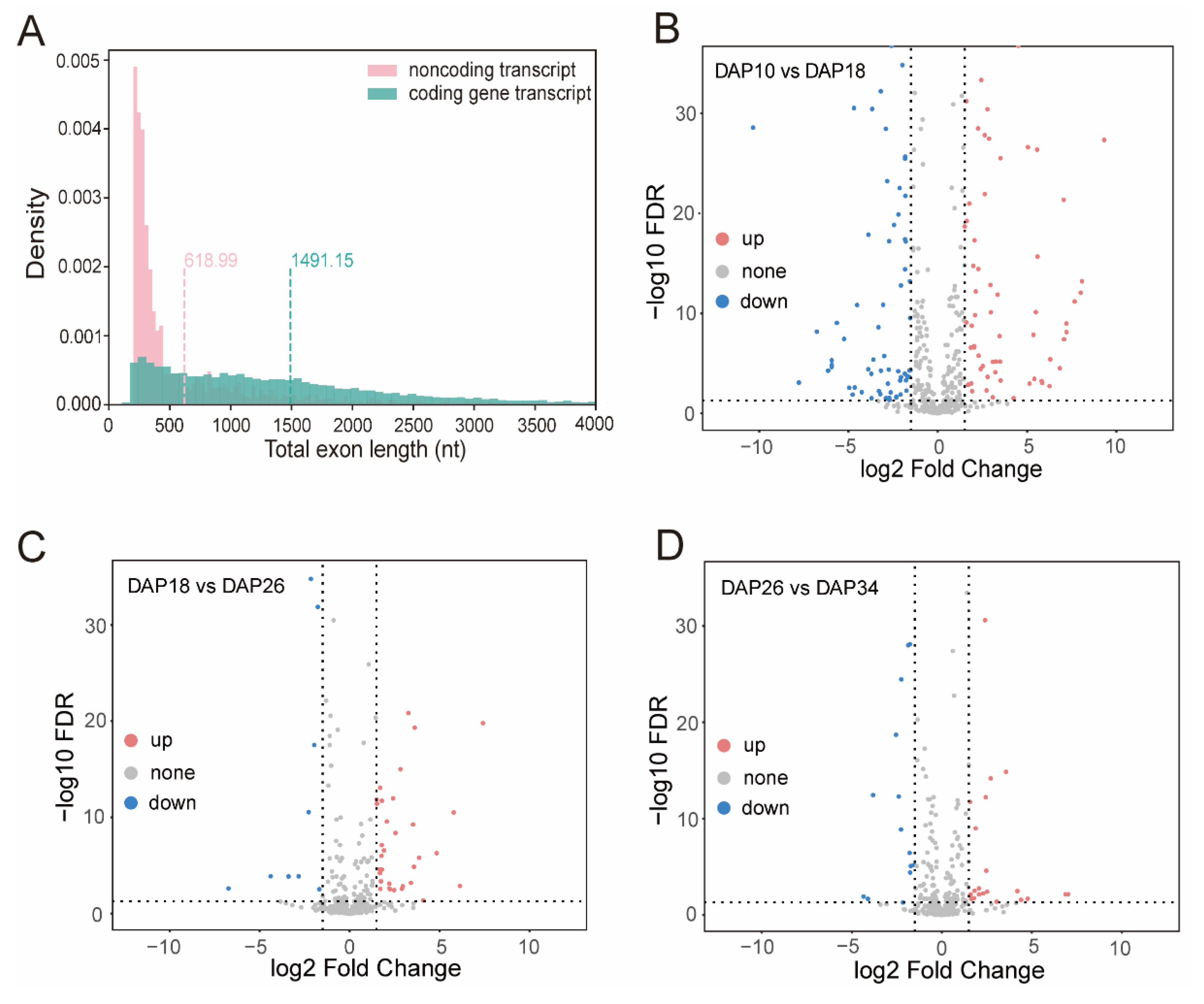
2.4. Identification of Long Non-Coding RNA from PacBio Sequences
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Total RNA Extraction, Construction of PacBio Library and Sequencing
4.3. Data Analysis
4.4. Quantification of Differential Alternative Splicing
4.5. Quantification of Differential Alternative Polyadenylation
4.6. Identification of lncRNA
4.7. PCR Validation of AS and APA
4.8. Data Presentation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Erickson, D.L.; Smith, B.D.; Clarke, A.C.; Sandweiss, D.H.; Tuross, N. An Asian origin for a 10,000-year-old domesticated plant in the Americas. Proc. Natl. Acad. Sci. USA 2005, 102, 18315–18320. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z.; et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 2013, 45, 51–58. [Google Scholar] [CrossRef]
- Giovannoni, J. Molecular Biology of Fruit Maturation and Ripening. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 725–749. [Google Scholar] [CrossRef]
- Seymour, G.B.; Østergaard, L.; Chapman, N.H.; Knapp, S.; Martin, C. Fruit development and ripening. Annu. Rev. Plant Biol. 2013, 64, 219–241. [Google Scholar] [CrossRef]
- Chen, T.; Qin, G.; Tian, S. Regulatory network of fruit ripening: Current understanding and future challenges. New Phytol. 2020, 228, 1219–1226. [Google Scholar] [CrossRef]
- Sánchez-Gómez, C.; Posé, D.; Martín-Pizarro, C. Insights into transcription factors controlling strawberry fruit development and ripening. Front. Plant Sci. 2022, 13, 1022369. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Chen, T.; Qin, G.; Tian, S. Current insights into posttranscriptional regulation of fleshy fruit ripening. Plant Physiol. 2023, 192, 1785–1798. [Google Scholar] [CrossRef]
- Tang, D.; Gallusci, P.; Lang, Z. Fruit development and epigenetic modifications. New Phytol. 2020, 228, 839–844. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, A. Recent advances in epigenetic triggering of climacteric fruit ripening. Plant Physiol. 2023, 192, 1711–1717. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.-K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef]
- Lu, T.; Lu, G.; Fan, D.; Zhu, C.; Li, W.; Zhao, Q.; Feng, Q.; Zhao, Y.; Guo, Y.; Li, W.; et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 2010, 20, 1238–1249. [Google Scholar] [CrossRef]
- Staiger, D.; Brown, J.W. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef]
- Wu, X.; Liu, M.; Downie, B.; Liang, C.; Ji, G.; Li, Q.Q.; Hunt, A.G. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. USA 2011, 108, 12533–12538. [Google Scholar] [CrossRef]
- Shen, Y.; Ji, G.; Haas, B.J.; Wu, X.; Zheng, J.; Reese, G.J.; Li, Q.Q. Genome level analysis of rice mRNA 3’-end processing signals and alternative polyadenylation. Nucleic Acids Res. 2008, 36, 3150–3161. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.; Zhang, H.; Wang, Y.; Lin, C.; Ma, L.; Gu, L. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 91, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Yuan, J.; Li, S.; Jia, S.; Han, L.; Xu, L. Analysis of transcripts and splice isoforms in red clover (Trifolium pratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, X.; Wang, L.; Wang, W.; Fu, C.; Yan, X.; Geng, X. A survey of transcriptome complexity using PacBio single-molecule real-time analysis combined with Illumina RNA sequencing for a better understanding of ricinoleic acid biosynthesis in Ricinus communis. BMC Genom. 2019, 20, 456. [Google Scholar] [CrossRef]
- Ye, J.; Cheng, S.; Zhou, X.; Chen, Z.; Kim, S.U.; Tan, J.; Zheng, J.; Xu, F.; Zhang, W.; Liao, Y.; et al. A global survey of full-length transcriptome of Ginkgo biloba reveals transcript variants involved in flavonoid biosynthesis. Ind. Crop. Prod. 2019, 139, 111547. [Google Scholar] [CrossRef]
- Chao, Q.; Gao, Z.F.; Zhang, D.; Zhao, B.G.; Dong, F.Q.; Fu, C.; Liu, L.; Wang, B. The developmental dynamics of the Populus stem transcriptome. Plant Biotechnol. J. 2019, 17, 206–219. [Google Scholar] [CrossRef]
- Deng, H.; Zhang, L.; Liao, M.; Wang, J.; Liang, D.; Xia, H.; Lv, X.; Deng, Q.; Wang, X.; Tang, Y.; et al. A PacBio single molecule real-time sequencing-based full-length transcriptome atlas of tree tomato (Solanum betaceum Cav.) and mining of simple sequence repeat markers. Front. Plant Sci. 2022, 13, 1052817. [Google Scholar] [CrossRef]
- Yu, Y.; Guo, S.; Ren, Y.; Zhang, J.; Li, M.; Tian, S.; Wang, J.; Sun, H.; Zuo, Y.; Chen, Y.; et al. Quantitative Transcriptomic and Proteomic Analysis of Fruit Development and Ripening in Watermelon (Citrullus lanatus). Front. Plant Sci. 2022, 13, 818392. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Haas, B.J.; Hamilton, J.P.; Mount, S.M.; Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 2006, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S. Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 2007, 58, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lv, W.; Shao, L.; Fu, Y.; Liu, H.; Yang, C.; Chen, A.; Xie, X.; Wang, Z.; Li, C. PacBio and Illumina RNA Sequencing Identify Alternative Splicing Events in Response to Cold Stress in Two Poplar Species. Front. Plant Sci. 2021, 12, 737004. [Google Scholar] [CrossRef]
- Xu, W.; Yang, T.; Wang, B.; Han, B.; Zhou, H.; Wang, Y.; Li, D.-Z.; Liu, A. Differential expression networks and inheritance patterns of long non-coding RNAs in castor bean seeds. Plant J. 2018, 95, 324–340. [Google Scholar] [CrossRef]
- Sun, Y.; Xiao, H. Identification of alternative splicing events by RNA sequencing in early growth tomato fruits. BMC Genom. 2015, 16, 948. [Google Scholar] [CrossRef]
- Tang, W.; Zheng, Y.; Dong, J.; Yu, J.; Yue, J.; Liu, F.; Guo, X.; Huang, S.; Wisniewski, M.; Sun, J.; et al. Comprehensive Transcriptome Profiling Reveals Long Noncoding RNA Expression and Alternative Splicing Regulation during Fruit Development and Ripening in Kiwifruit (Actinidia chinensis). Front. Plant Sci. 2016, 7, 335. [Google Scholar] [CrossRef]
- Guodong, R.; Jianguo, Z.; Xiaoxia, L.; Ying, L. Identification of putative genes for polyphenol biosynthesis in olive fruits and leaves using full-length transcriptome sequencing. Food Chem. 2019, 300, 125246. [Google Scholar] [CrossRef]
- Yan, X.; Bai, D.; Song, H.; Lin, K.; Pang, E. Alternative splicing during fruit development among fleshy fruits. BMC Genom. 2021, 22, 762. [Google Scholar] [CrossRef]
- Gupta, V.; Estrada, A.D.; Blakley, I.; Reid, R.; Patel, K.; Meyer, M.D.; Andersen, S.U.; Brown, A.F.; Lila, M.A.; Loraine, A.E. RNA-Seq analysis and annotation of a draft blueberry genome assembly identifies candidate genes involved in fruit ripening, biosynthesis of bioactive compounds, and stage-specific alternative splicing. Gigascience 2015, 4, 5. [Google Scholar] [CrossRef]
- Terol, J.; Tadeo, F.; Ventimilla, D.; Talon, M. An RNA-Seq-based reference transcriptome for Citrus. Plant Biotechnol. J. 2016, 14, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, X.; Liu, C.; Liu, G.; Li, S.; Wang, L. Integrating Omics and Alternative Splicing Reveals Insights into Grape Response to High Temperature. Plant Physiol. 2017, 173, 1502–1518. [Google Scholar] [CrossRef]
- Xanthopoulou, A.; Moysiadis, T.; Bazakos, C.; Karagiannis, E.; Karamichali, I.; Stamatakis, G.; Samiotaki, M.; Manioudaki, M.; Michailidis, M.; Madesis, P.; et al. The perennial fruit tree proteogenomics atlas: A spatial map of the sweet cherry proteome and transcriptome. Plant J. 2022, 109, 1319–1336. [Google Scholar] [CrossRef]
- Yuan, H.; Yu, H.; Huang, T.; Shen, X.; Xia, J.; Pang, F.; Wang, J.; Zhao, M. The complexity of the Fragaria x ananassa (octoploid) transcriptome by single-molecule long-read sequencing. Hortic. Res. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Saminathan, T.; Nimmakayala, P.; Manohar, S.; Malkaram, S.; Almeida, A.; Cantrell, R.; Tomason, Y.; Abburi, L.; Rahman, M.A.; Vajja, V.G.; et al. Differential gene expression and alternative splicing between diploid and tetraploid watermelon. J. Exp. Bot. 2015, 66, 1369–1385. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Yang, D.; Su, W.; Ma, L.; Shen, Y.; Ji, G.; Ye, X.; Wu, X.; Li, Q.Q. Genome-wide dynamics of alternative polyadenylation in rice. Genome Res. 2016, 26, 1753–1760. [Google Scholar] [CrossRef]
- Sherstnev, A.; Duc, C.; Cole, C.; Zacharaki, V.; Hornyik, C.; Ozsolak, F.; Milos, P.M.; Barton, G.J.; Simpson, G.G. Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat. Struct. Mol. Biol. 2012, 19, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; La, H.; Lu, K.; Wang, P.; Miki, D.; Ren, Z.; Duan, C.-G.; Wang, X.; Tang, K.; Zeng, L.; et al. Arabidopsis EDM2 promotes IBM1 distal polyadenylation and regulates genome DNA methylation patterns. Proc. Natl. Acad. Sci. USA 2014, 111, 527–532. [Google Scholar] [CrossRef]
- Wang, X.; Duan, C.G.; Tang, K.; Wang, B.; Zhang, H.; Lei, M.; Lu, K.; Mangrauthia, S.K.; Wang, P.; Zhu, G.; et al. RNA-binding protein regulates plant DNA methylation by controlling mRNA processing at the intronic heterochromatin-containing gene IBM1. Proc. Natl. Acad. Sci. USA 2013, 110, 15467–15472. [Google Scholar] [CrossRef]
- Au, K.F.; Underwood, J.G.; Lee, L.; Wong, W.H. Improving PacBio long read accuracy by short read alignment. PLoS ONE 2012, 7, e46679. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | cDNAsize | Number of Reads of Insert | Number of Five-Five Primer Reads | Number of Three-Three Primber Reads | Number of Adaptor Reads | Number of Filtered Short Reads | Number of Full-Length Non-Chimeric Reads | Error-Corrected and Mapped FLNC |
|---|---|---|---|---|---|---|---|---|
| DAP10-1 | 1-10k | 632,678 | 19,259 | 7905 | 3 | 0 | 580,464 | 577,193 |
| DAP10-2 | 1-10k | 726,681 | 12,245 | 22,527 | 12 | 0 | 663,057 | 661,727 |
| DAP10-3 | 1-10k | 639,781 | 146,488 | 7345 | 18 | 0 | 463,837 | 460,828 |
| DAP18-1 | 1-10k | 727,467 | 20,973 | 7599 | 14 | 0 | 681,511 | 679,326 |
| DAP18-2 | 1-10k | 696,383 | 104,864 | 12,243 | 9 | 0 | 557,964 | 555,213 |
| DAP18-3 | 1-10k | 690,537 | 29,087 | 11,170 | 15 | 0 | 629,609 | 626,298 |
| DAP26-1 | 1-10k | 618,696 | 21,428 | 12,025 | 6 | 0 | 573,505 | 569,181 |
| DAP26-2 | 1-10k | 707,380 | 24,627 | 14,069 | 7 | 0 | 654,125 | 651,197 |
| DAP26-3 | 1-10k | 550,489 | 13,507 | 5581 | 7 | 0 | 520,269 | 516,792 |
| DAP34-1 | 1-10k | 587,052 | 98,357 | 18,328 | 42 | 0 | 457,391 | 453,471 |
| DAP34-2 | 1-10k | 531,201 | 11,605 | 5697 | 3 | 0 | 502,478 | 499,375 |
| DAP34-3 | 1-10k | 729,105 | 27,788 | 14,078 | 3 | 0 | 673,739 | 670,694 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Liufu, Y.; Ren, Y.; Zhang, J.; Li, M.; Tian, S.; Wang, J.; Liao, S.; Gong, G.; Zhang, H.; et al. Comprehensive Profiling of Alternative Splicing and Alternative Polyadenylation during Fruit Ripening in Watermelon (Citrullus lanatus). Int. J. Mol. Sci. 2023, 24, 15333. https://doi.org/10.3390/ijms242015333
Yu Y, Liufu Y, Ren Y, Zhang J, Li M, Tian S, Wang J, Liao S, Gong G, Zhang H, et al. Comprehensive Profiling of Alternative Splicing and Alternative Polyadenylation during Fruit Ripening in Watermelon (Citrullus lanatus). International Journal of Molecular Sciences. 2023; 24(20):15333. https://doi.org/10.3390/ijms242015333
Chicago/Turabian StyleYu, Yongtao, Yuxiang Liufu, Yi Ren, Jie Zhang, Maoying Li, Shouwei Tian, Jinfang Wang, Shengjin Liao, Guoyi Gong, Haiying Zhang, and et al. 2023. "Comprehensive Profiling of Alternative Splicing and Alternative Polyadenylation during Fruit Ripening in Watermelon (Citrullus lanatus)" International Journal of Molecular Sciences 24, no. 20: 15333. https://doi.org/10.3390/ijms242015333
APA StyleYu, Y., Liufu, Y., Ren, Y., Zhang, J., Li, M., Tian, S., Wang, J., Liao, S., Gong, G., Zhang, H., & Guo, S. (2023). Comprehensive Profiling of Alternative Splicing and Alternative Polyadenylation during Fruit Ripening in Watermelon (Citrullus lanatus). International Journal of Molecular Sciences, 24(20), 15333. https://doi.org/10.3390/ijms242015333