

Transcriptome and Physiological Analysis of Rapeseed Tolerance to Post-Flowering Temperature Increase




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effects of Heat Stress on Seed Yield and Quality Traits of Two Rapeseed Genotypes
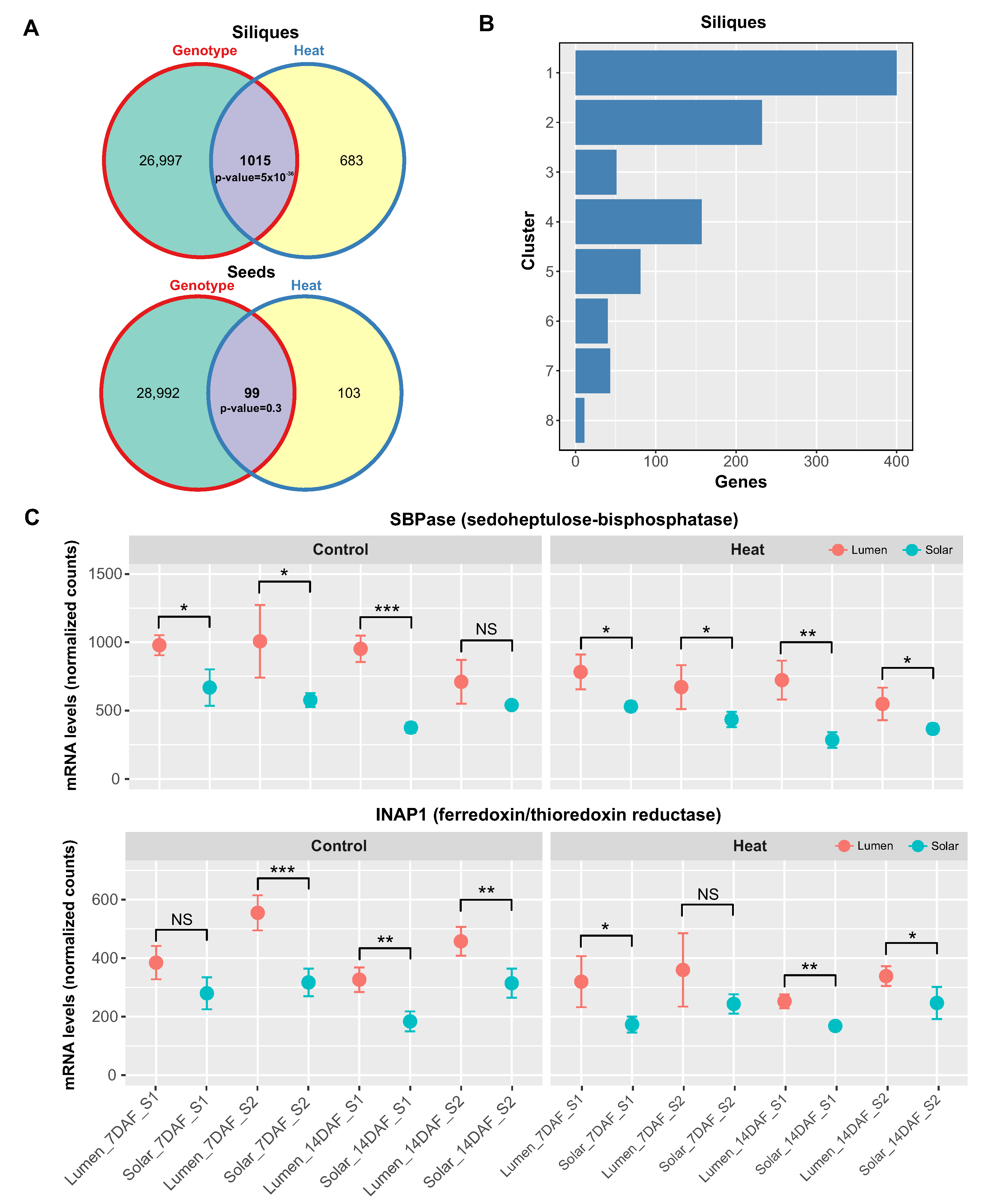
2.2. Multivariate Analysis of RNA-Seq Data Reveals Differential Effect of Heat Stress on Source and Sink Tissues
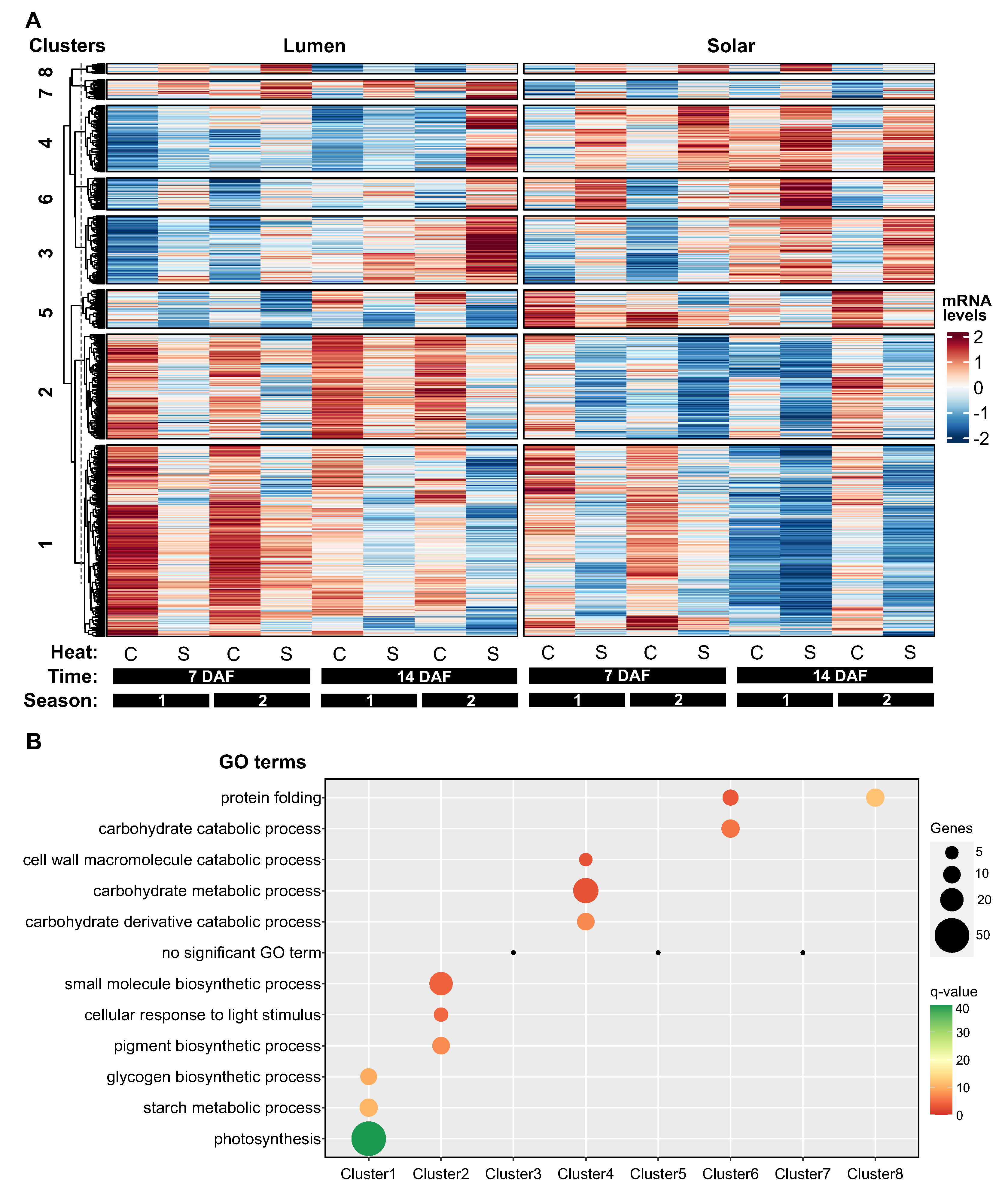
2.3. Hierarchical Clustering Unveils Patterns of Heat-Induced Gene Expression in Siliques
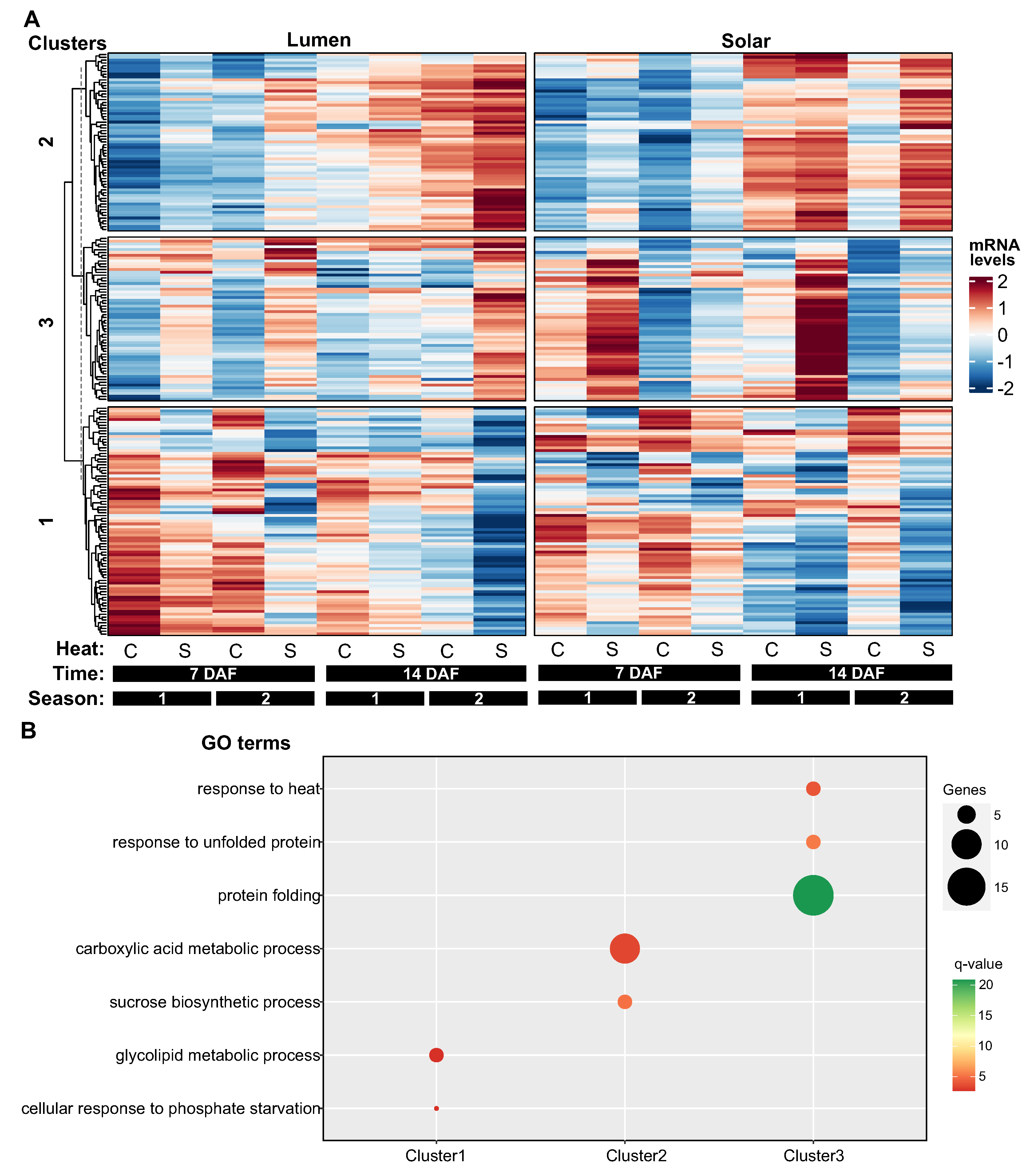
2.4. Hierarchical Clustering Reveals Three Distinct Patterns of Heat-Responsive Gene Expression in Seeds
2.5. Genotype-Specific Effects of Heat Stress on Photosynthesis-Related Genes in Siliques
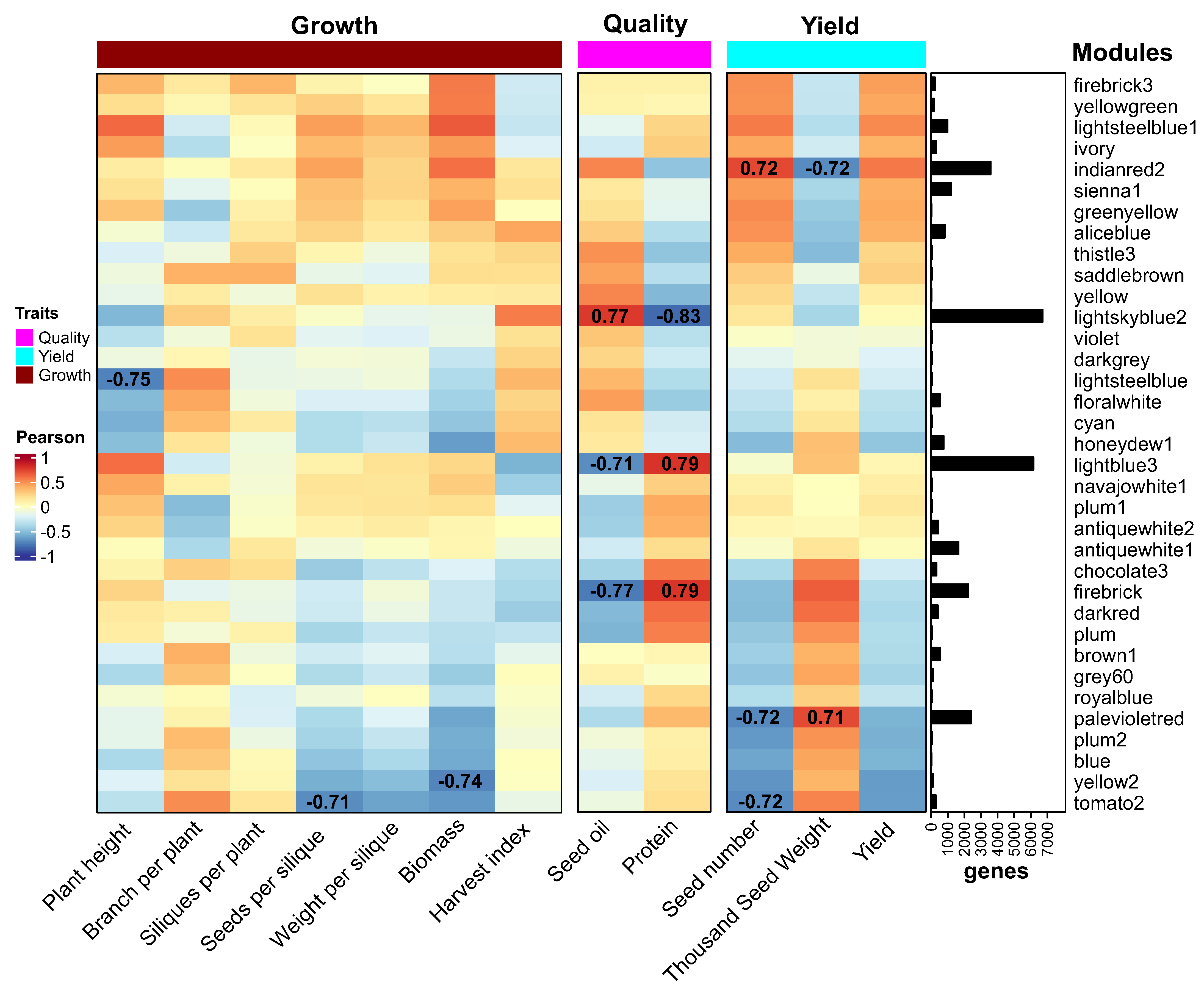
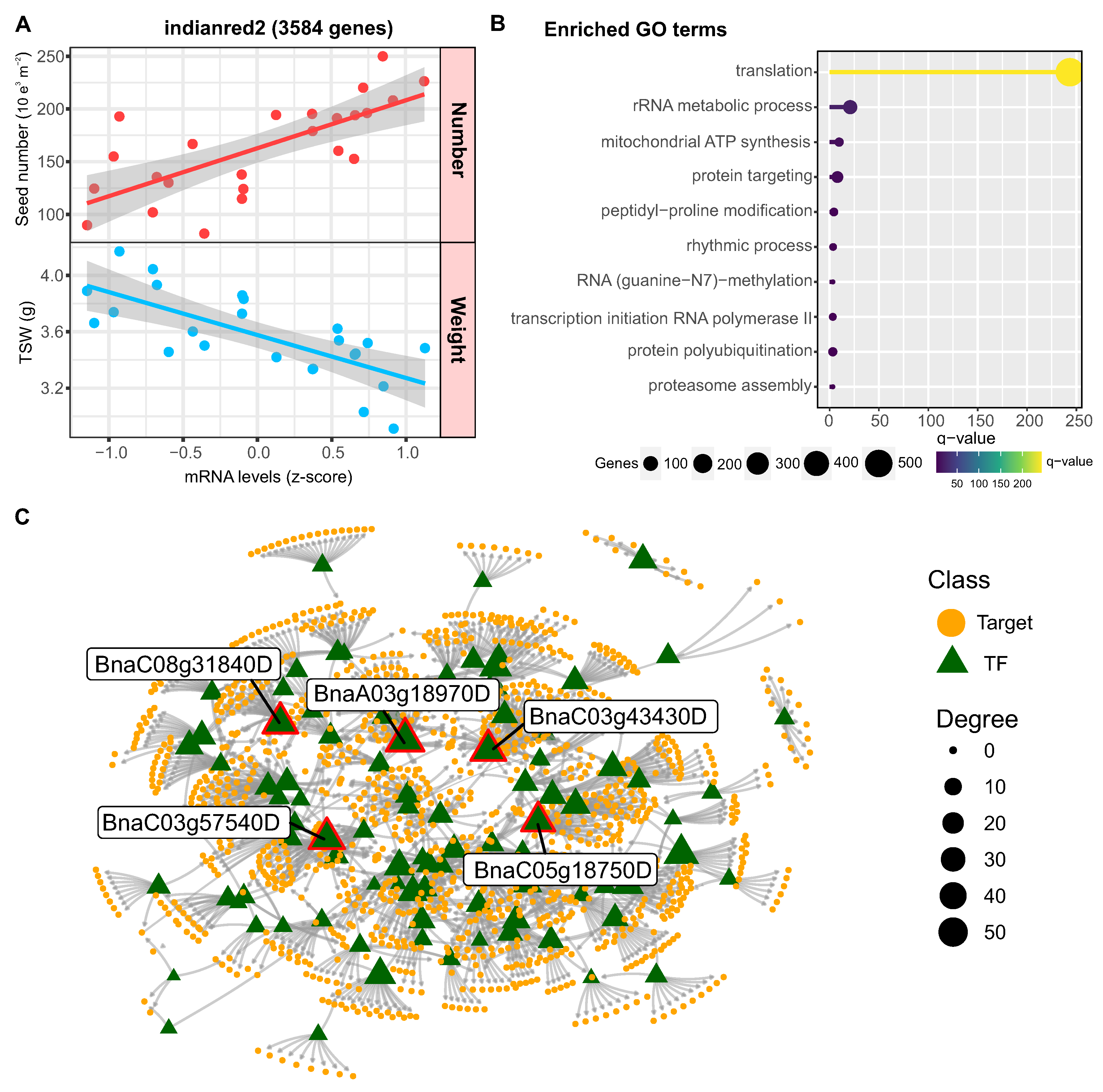
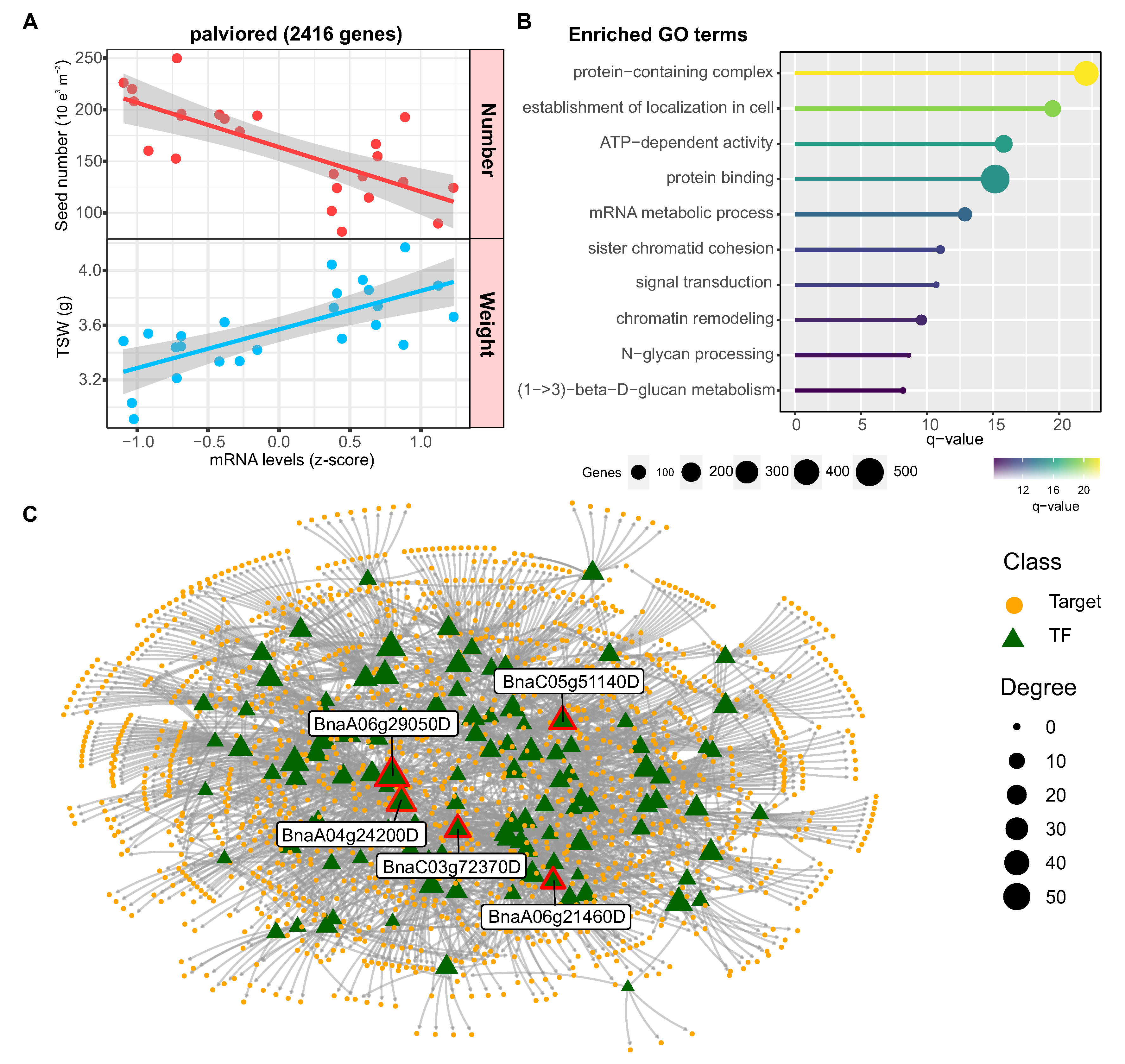
2.6. Co-Expression Network Analysis Reveals Major Modules for Rapeseed Yield and Quality
2.7. Identification of Regulatory Factors Associated with the Compensation of SW and SN Flowering
3. Discussion
3.1. Divergent Molecular Responses to Heat Stress in Source and Sink Tissues
3.2. Differential Transcriptome Rapeseed Genotype Responses to Heat Stress
3.3. Distinct Regulators Mediate the Seed Number versus Weight Trade-Off
4. Materials and Methods
4.1. Field Experiment, Treatments, and Crop Management
4.2. Phenology and Physiological Plant Sampling
4.3. Seed Sampling and RNA Isolation
4.4. RNA-Seq Analysis
4.5. Hierarchical Clustering Analysis
4.6. Gene Coexpression Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, W.; Sakai, H.; Yagi, K.; Hasegawa, T. Interactions of elevated [CO2] and night temperature on rice growth and yield. Agric. For. Meteorol. 2009, 149, 51–58. [Google Scholar] [CrossRef]
- Kim, H.R.; You, Y.H. Effects of elevated CO2 concentration and increased temperature on leaf related-physiological responses of Phytolacca insularis (native species) and Phytolacca americana (invasive species). J. Ecol. Environ. 2010, 33, 195–204. [Google Scholar] [CrossRef]
- Ray, D.K.; West, P.C.; Clark, M.; Gerber, J.S.; Prishchepov, A.V.; Chatterjee, S. Climate change has likely already affected global food production. PLoS ONE 2019, 14, e0217148. [Google Scholar] [CrossRef] [PubMed]
- IPCC. Climate Change 2021—The Physical Science Basis: Working Group I Contribution to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2023. [Google Scholar] [CrossRef]
- Rivelli, G.M.; Long, M.E.F.; Abeledo, L.G.; Calderini, D.F.; Miralles, D.J.; Rondanini, D.P. Assessment of heat stress and cloudiness probabilities in post-flowering of spring wheat and canola in the Southern Cone of South America. Theor. Appl. Climatol. 2021, 145, 1485–1502. [Google Scholar] [CrossRef]
- Sandaña, P.A.; Harcha, C.I.; Calderini, D.F. Sensitivity of yield and grain nitrogen concentration of wheat lupin and pea to source reduction during grain filling. A comparative survey under high yielding conditions. Field Crops Res. 2009, 114, 233–243. [Google Scholar] [CrossRef]
- Mera, M.; Lizana, X.C.; Calderini, D.F. Cropping systems in environments with high yield potential of southern Chile. In Crop Physiology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 111–140. [Google Scholar] [CrossRef]
- Verdejo, J.; Calderini, D.F. Plasticity of seed weight in winter and spring rapeseed is higher in a narrow but different window after flowering. Field Crops Res. 2020, 250, 107777. [Google Scholar] [CrossRef]
- del Pozo, A.; Jobet, C.; Matus, I.; Méndez-Espinoza, A.M.; Garriga, M.; Castillo, D.; Elazab, A. Genetic Yield Gains and Changes in Morphophysiological-Related Traits of Winter Wheat in Southern Chilean High-Yielding Environments. Front. Plant Sci. 2022, 12, 732988. [Google Scholar] [CrossRef]
- Slafer, G.; Rawson, H. Sensitivity of Wheat Phasic Development to Major Environmental Factors: A Re-Examination of Some Assumptions Made by Physiologists and Modellers. Funct. Plant Biol. 1994, 21, 393. [Google Scholar] [CrossRef]
- Sadras, V.O.; Calderini, D.F. Crop Physiology Case Histories for Major Crops; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Secchi, M.A.; Fernandez, J.A.; Stamm, M.J.; Durrett, T.; Prasad, P.V.; Messina, C.D.; Ciampitti, I.A. Effects of heat and drought on canola (Brassica napus L.) yield oil, and protein: A meta-analysis. Field Crops Res. 2023, 293, 108848. [Google Scholar] [CrossRef]
- Kourani, M.; Mohareb, F.; Rezwan, F.I.; Anastasiadi, M.; Hammond, J.P. Genetic and Physiological Responses to Heat Stress in Brassica napus. Front. Plant Sci. 2022, 13, 832147. [Google Scholar] [CrossRef]
- Young, L.W.; Wilen, R.W.; Bonham-Smith, P.C. High temperature stress of Brassica napus during flowering reduces micro- and megagametophyte fertility induces fruit abortion, and disrupts seed production. J. Exp. Bot. 2004, 55, 485–495. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, L.M.M.; Coquemont-Guyot, M.; Elie, N.; Morvan-Bertrand, A.; Avice, J.C.; Mollier, A.; Brunel-Muguet, S. Repeated heat stress events during the reproductive phase impact the dynamic development of seeds in Brassica napus L. Plant Sci. 2023, 327, 111559. [Google Scholar] [CrossRef]
- Chen, S.; Stefanova, K.; Siddique, K.H.M.; Cowling, W.A. Transient daily heat stress during the early reproductive phase disrupts pod and seed development in Brassica napus L. Food Energy Secur. 2020, 10, e262. [Google Scholar] [CrossRef]
- Jedličková, V.; Hejret, V.; Demko, M.; Jedlička, P.; Štefková, M.; Robert, H.S. Transcriptome analysis of thermomorphogenesis in ovules and during early seed development in Brassica napus. BMC Genom. 2023, 24, 236. [Google Scholar] [CrossRef]
- Gao, G.; Hu, J.; Zhang, X.; Zhang, F.; Li, M.; Wu, X. Transcriptome analysis reveals genes expression pattern of seed response to heat stress in Brassica napus L. Oil Crop Sci. 2021, 6, 87–96. [Google Scholar] [CrossRef]
- Lohani, N.; Singh, M.B.; Bhalla, P.L. RNA-Seq Highlights Molecular Events Associated With Impaired Pollen-Pistil Interactions Following Short-Term Heat Stress in Brassica napus. Front. Plant Sci. 2021, 11, 622748. [Google Scholar] [CrossRef] [PubMed]
- Canales, J.; Verdejo, J.; Carrasco-Puga, G.; Castillo, F.M.; Arenas-M, A.; Calderini, D.F. Transcriptome Analysis of Seed Weight Plasticity in Brassica napus. Int. J. Mol. Sci. 2021, 22, 4449. [Google Scholar] [CrossRef]
- Li, N.; Song, D.; Peng, W.; Zhan, J.; Shi, J.; Wang, X.; Liu, G.; Wang, H. Maternal control of seed weight in rapeseed (Brassica napus L.): The causal link between the size of pod (mother source) and seed (offspring, sink). Plant Biotechnol. J. 2018, 17, 736–749. [Google Scholar] [CrossRef]
- Dong, H.; Tan, C.; Li, Y.; He, Y.; Wei, S.; Cui, Y.; Chen, Y.; Wei, D.; Fu, Y.; He, Y.; et al. Genome-Wide Association Study Reveals Both Overlapping and Independent Genetic Loci to Control Seed Weight and Silique Length in Brassica napus. Front. Plant Sci. 2018, 9, 921. [Google Scholar] [CrossRef]
- Kirkegaard, J.A.; Lilley, J.M.; Brill, R.D.; Ware, A.H.; Walela, C.K. The critical period for yield and quality determination in canola (Brassica napus L.). Field Crops Res. 2018, 222, 180–188. [Google Scholar] [CrossRef]
- Labra, M.H.; Struik, P.C.; Evers, J.B.; Calderini, D.F. Plasticity of seed weight compensates reductions in seed number of oilseed rape in response to shading at flowering. Eur. J. Agron. 2017, 84, 113–124. [Google Scholar] [CrossRef]
- Rivelli, G.; Calderini, D.F.; Abeledo, L.G.; Miralles, D.J.; Rondanini, D.P. Yield and Quality Traits of Wheat and Rapeseed in Response to Source-Sink Ratio and Heat Stress in Post-Flowering. Soc. Sci. Res. Netw. 2023. [Google Scholar] [CrossRef]
- Gu, Z. Complex heatmap visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Liu, X.; Yu, H.; Guan, Y.; Li, J.; Guo, F. Carbonylation and loss-of-function analyses of SBPase reveal its metabolic interface role in oxidative stress, carbon assimilation, and multiple aspects of growth and development in Arabidopsis. Mol. Plant 2012, 5, 1082–1099. [Google Scholar] [CrossRef]
- Hashida, S.; Miyagi, A.; Nishiyama, M.; Yoshida, K.; Hisabori, T.; Kawai-Yamada, M. Ferredoxin/thioredoxin system plays an important role in the chloroplastic NADP status of Arabidopsis. Plant J. 2018, 95, 947–960. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Yu, H.; Murchie, E.H.; González-Carranza, Z.H.; Pyke, K.A.; Roberts, J.A. Decreased photosynthesis in the erect panicle 3 (ep3) mutant of rice is associated with reduced stomatal conductance and attenuated guard cell development. J. Exp. Bot. 2015, 66, 1543–1552. [Google Scholar] [CrossRef]
- Villanueva, J.M.; Broadhvest, J.; Hauser, B.A.; Meister, R.J.; Schneitz, K.; Gasser, C.S. INNER NO OUTER regulates abaxial- adaxial patterning in Arabidopsis ovules. Genes Dev. 1999, 13, 3160–3169. [Google Scholar] [CrossRef]
- Shu, K.; Chen, Q.; Wu, Y.; Liu, R.; Zhang, H.; Wang, P.; Li, Y.; Wang, S.; Tang, S.; Liu, C.; et al. ABI4 mediates antagonistic effects of abscisic acid and gibberellins at transcript and protein levels. Plant J. 2016, 85, 348–361. [Google Scholar] [CrossRef]
- Renard, J.; Niñoles, R.; Martínez-Almonacid, I.; Gayubas, B.; Mateos-Fernández, R.; Bissoli, G.; Bueso, E.; Serrano, R.; Gadea, J. Identification of novel seed longevity genes related to oxidative stress and seed coat by genome-wide association studies and reverse genetics. Plant Cell Environ. 2020, 43, 2523–2539. [Google Scholar] [CrossRef]
- Schruff, M.C.; Spielman, M.; Tiwari, S.; Adams, S.; Fenby, N.; Scott, R.J. The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signalling, cell division, and the size of seeds and other organs. Development 2006, 133, 251–261. [Google Scholar] [CrossRef]
- Angadi, S.V.; Cutforth, H.W.; Miller, P.R.; McConkey, B.G.; Entz, M.H.; Brandt, S.A.; Volkmar, K.M. Response of three Brassica species to high temperature stress during reproductive growth. Can. J. Plant Sci. 2000, 80, 693–701. [Google Scholar] [CrossRef]
- Lohani, N.; Singh, M.B.; Bhalla, P.L. High temperature susceptibility of sexual reproduction in crop plants. J. Exp. Bot. 2019, 71, 555–568. [Google Scholar] [CrossRef]
- Mácová, K.; Prabhullachandran, U.; Štefková, M.; Spyroglou, I.; Pěnčík, A.; Endlová, L.; Novák, O.; Robert, H.S. Long-Term High-Temperature Stress Impacts on Embryo and Seed Development in Brassica napus. Front. Plant Sci. 2022, 13. [Google Scholar] [CrossRef]
- Kim, K.; Portis, A.R. Temperature Dependence of Photosynthesis in Arabidopsis Plants with Modifications in Rubisco Activase and Membrane Fluidity. Plant Cell Physiol. 2005, 46, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, K.B.; Lu, Z.G.; Ren, S.X.; Jiang, H.R.; Cui, J.W.; Chen, G.; Teng, N.J.; Lam, H.M.; Jin, B. Differential physiological transcriptomic and metabolomic responses of Arabidopsis leaves under prolonged warming and heat shock. BMC Plant Biol. 2020, 20, 86. [Google Scholar] [CrossRef]
- Huang, R.; Liu, Z.; Xing, M.; Yang, Y.; Wu, X.; Liu, H.; Liang, W. Heat Stress Suppresses Brassica napus Seed Oil Accumulation by Inhibition of Photosynthesis and BnWRI1 Pathway. Plant Cell Physiol. 2019, 60, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- ul Haq, S.; Khan, A.; Ali, M.; Khattak, A.M.; Gai, W.X.; Zhang, H.X.; Wei, A.M.; Gong, Z.H. Heat Shock Proteins: Dynamic Biomolecules to Counter Plant Biotic and Abiotic Stresses. Int. J. Mol. Sci. 2019, 20, 5321. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Araki, M.; Okamura, K.; Ishibashi, Y.; Yuasa, T.; Iwaya-Inoue, M. Assimilate translocation and expression of sucrose transporter OsSUT1, contribute to high-performance ripening under heat stress in the heat-tolerant rice cultivar Genkitsukushi. J. Plant Physiol. 2013, 170, 1579–1584. [Google Scholar] [CrossRef]
- Moore, C.E.; Meacham-Hensold, K.; Lemonnier, P.; Slattery, R.A.; Benjamin, C.; Bernacchi, C.J.; Lawson, T.; Cavanagh, A.P. The effect of increasing temperature on crop photosynthesis: From enzymes to ecosystems. J. Exp. Bot. 2021, 72, 2822–2844. [Google Scholar] [CrossRef]
- Abdelrahman, M.; Burritt, D.J.; Gupta, A.; Tsujimoto, H.; Tran, L.S.P. Heat stress effects on source–sink relationships and metabolome dynamics in wheat. J. Exp. Bot. 2019, 71, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bhushan, B.; Wakchaure, G.C.; Dutta, R.; Jat, B.S.; Meena, K.K.; Rakshit, S.; Pathak, H. Unveiling the impact of heat stress on seed biochemical composition of major cereal crops: Implications for crop resilience and nutritional value. Plant Stress 2023, 9, 100183. [Google Scholar] [CrossRef]
- Harrison, E.P.; Olcer, H.; Lloyd, J.C.; Long, S.P.; Raines, C.A. Small decreases in SBPase cause a linear decline in the apparent RuBP regeneration rate but do not affect Rubisco carboxylation capacity. J. Exp. Bot. 2001, 52, 1779–1784. [Google Scholar] [CrossRef]
- Harrison, E.P.; Willingham, N.M.; Lloyd, J.C.; Raines, C.A. Reduced sedoheptulose-1,7-bisphosphatase levels in transgenic tobacco lead to decreased photosynthetic capacity and altered carbohydrate accumulation. Planta 1997, 204, 27–36. [Google Scholar] [CrossRef]
- Lawson, T.; Bryant, B.; Lefebvre, S.; Lloyd, J.C.; Raines, C.A. Decreased SBPase activity alters growth and development in transgenic tobacco plants. Plant Cell Environ. 2006, 29, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, Y.; Tamoi, M.; Shigeoka, S. Overexpression of a cyanobacterial fructose-1,6-/sedoheptulose-1,7-bisphosphatase in tobacco enhances photosynthesis and growth. Nat. Biotechnol. 2001, 19, 965–969. [Google Scholar] [CrossRef]
- Lefebvre, S.; Lawson, T.; Fryer, M.; Zakhleniuk, O.V.; Lloyd, J.C.; Raines, C.A. Increased Sedoheptulose-1,7-Bisphosphatase Activity in Transgenic Tobacco Plants Stimulates Photosynthesis and Growth from an Early Stage in Development. Plant Physiol. 2005, 138, 451–460. [Google Scholar] [CrossRef]
- Skinner, D.J.; Dang, T.; Gasser, C.S. The Arabidopsis INNER NO OUTER (INO) gene acts exclusively and quantitatively in regulation of ovule outer integument development. Plant Direct 2023, 7, e485. [Google Scholar] [CrossRef]
- Skinner, D.J. Regulation of Ovule Development. Plant Cell Online 2004, 16, S32–S45. [Google Scholar] [CrossRef]
- Jia, D.; Chen, L.G.; Yin, G.; Yang, X.; Gao, Z.; Guo, Y.; Sun, Y.; Tang, W. Brassinosteroids regulate outer ovule integument growth in part via the control of INNER NO OUTER by BRASSINOZOLE-RESISTANT family transcription factors. J. Integr. Plant Biol. 2020, 62, 1093–1111. [Google Scholar] [CrossRef] [PubMed]
- Adamski, N.M.; Anastasiou, E.; Eriksson, S.; O’Neill, C.M.; Lenhard, M. Local maternal control of seed size by KLUH/CYP78A5-dependent growth signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 20115–20120. [Google Scholar] [CrossRef]
- Meier, U. Growth Stages of Mono-And Dicotyledonous Plants; Julius Kühn-Institut: Quedlinburg, Germany, 2018. [Google Scholar]
- Kirk, P.L. Kjeldahl Method for Total Nitrogen. Anal. Chem. 1950, 22, 354–358. [Google Scholar] [CrossRef]
- Merrill, A.; Watt, B. Energy Value of Foods: Basis and Derivation; Scientific Research Publishing: Wuhan, China, 1973. [Google Scholar]
- Graeber, K.; Linkies, A.; Wood, A.T.; Leubner-Metzger, G. A Guideline to Family-Wide Comparative State-of-the-Art Quantitative RT-PCR Analysis Exemplified with a Brassicaceae Cross-Species Seed Germination Case Study. Plant Cell 2011, 23, 2045–2063. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, H.; Buxdorf, K.; Shapira, I.; Ein-Gedi, S.; Zvi, M.M.B.; Fridman, E.; Moshelion, M.; Levy, M. LogSpin: A simple economical and fast method for RNA isolation from infected or healthy plants and other eukaryotic tissues. BMC Res. Notes 2012, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 2007, 24, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 2020, 9, 709. [Google Scholar] [CrossRef]
- Almeida-Silva, F.; Venancio, T.M. BioNERO: An all-in-one R/Bioconductor package for comprehensive and easy biological network reconstruction. Funct. Integr. Genom. 2021, 22, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-Scale Mapping and Validation of Escherichia coli Transcriptional Regulation from a Compendium of Expression Profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Lizana, X.C.; Calderini, D.F. Yield and grain quality of wheat in response to increased temperatures at key periods for grain number and grain weight determination: Considerations for the climatic change scenarios of Chile. J. Agric. Sci. 2013, 151, 209–221. [Google Scholar] [CrossRef]
- Kutner, M.; Nachtsheim, C.; Neter, J. Applied Linear Regression Models; McGraw-Hill Education: Boston, MA, USA, 2004; p. 701. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canales, J.; Verdejo, J.F.; Calderini, D.F. Transcriptome and Physiological Analysis of Rapeseed Tolerance to Post-Flowering Temperature Increase. Int. J. Mol. Sci. 2023, 24, 15593. https://doi.org/10.3390/ijms242115593
Canales J, Verdejo JF, Calderini DF. Transcriptome and Physiological Analysis of Rapeseed Tolerance to Post-Flowering Temperature Increase. International Journal of Molecular Sciences. 2023; 24(21):15593. https://doi.org/10.3390/ijms242115593
Chicago/Turabian StyleCanales, Javier, José F. Verdejo, and Daniel F. Calderini. 2023. "Transcriptome and Physiological Analysis of Rapeseed Tolerance to Post-Flowering Temperature Increase" International Journal of Molecular Sciences 24, no. 21: 15593. https://doi.org/10.3390/ijms242115593
APA StyleCanales, J., Verdejo, J. F., & Calderini, D. F. (2023). Transcriptome and Physiological Analysis of Rapeseed Tolerance to Post-Flowering Temperature Increase. International Journal of Molecular Sciences, 24(21), 15593. https://doi.org/10.3390/ijms242115593