
Inflammatory Cytokine Elaboration Following Secondhand Smoke (SHS) Exposure Is Mediated in Part by RAGE Signaling


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
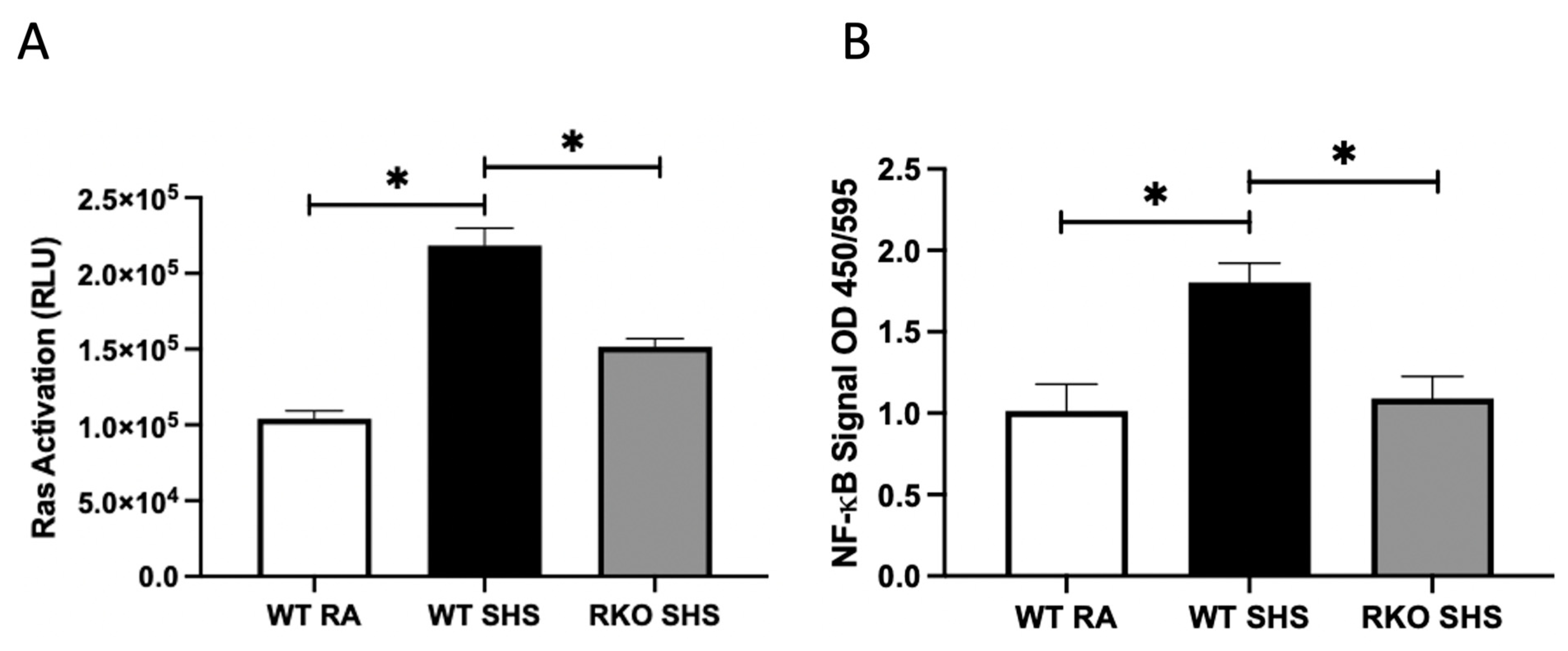
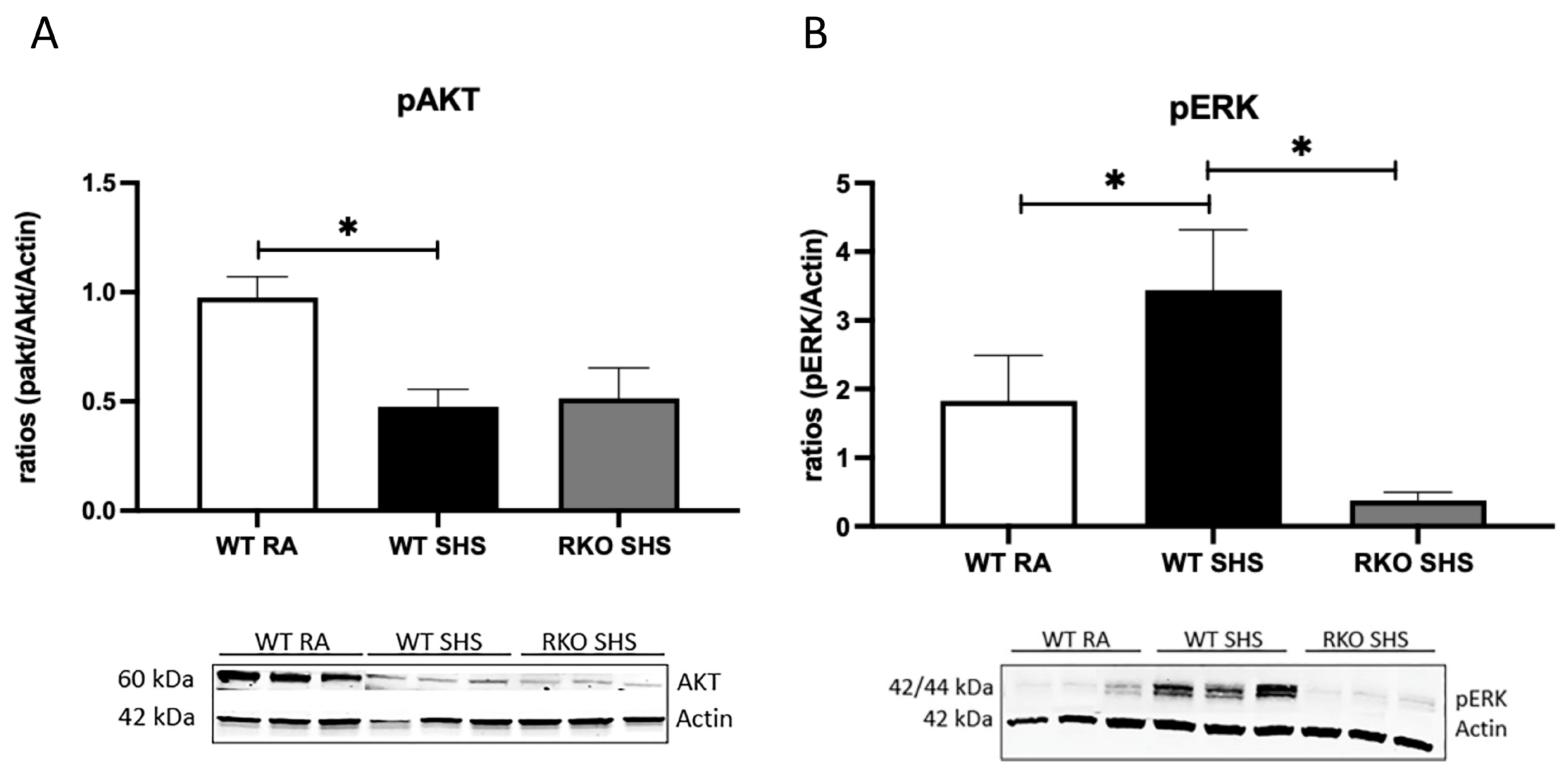
2.1. Cellular Signaling Pathways Following Chronic SHS Exposure
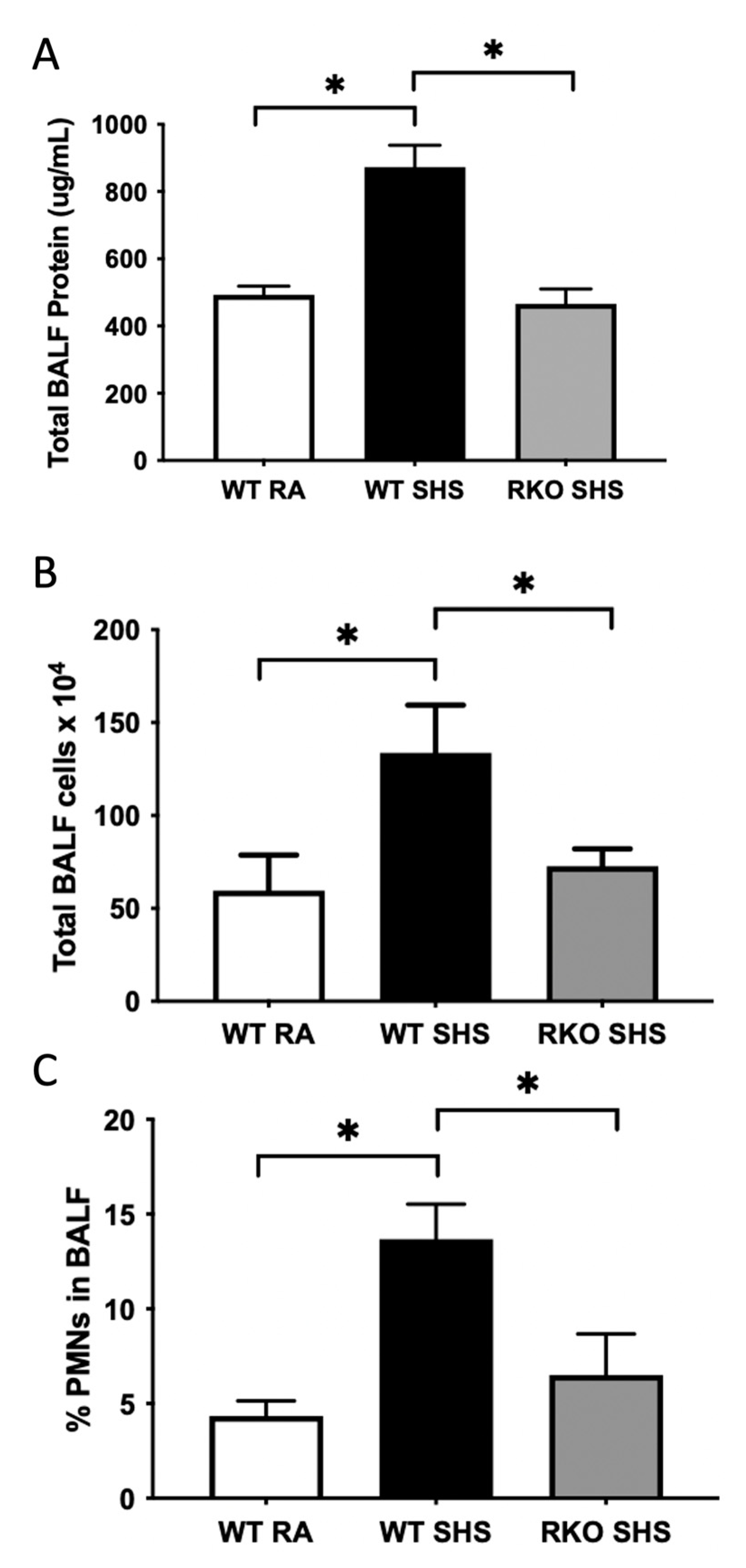
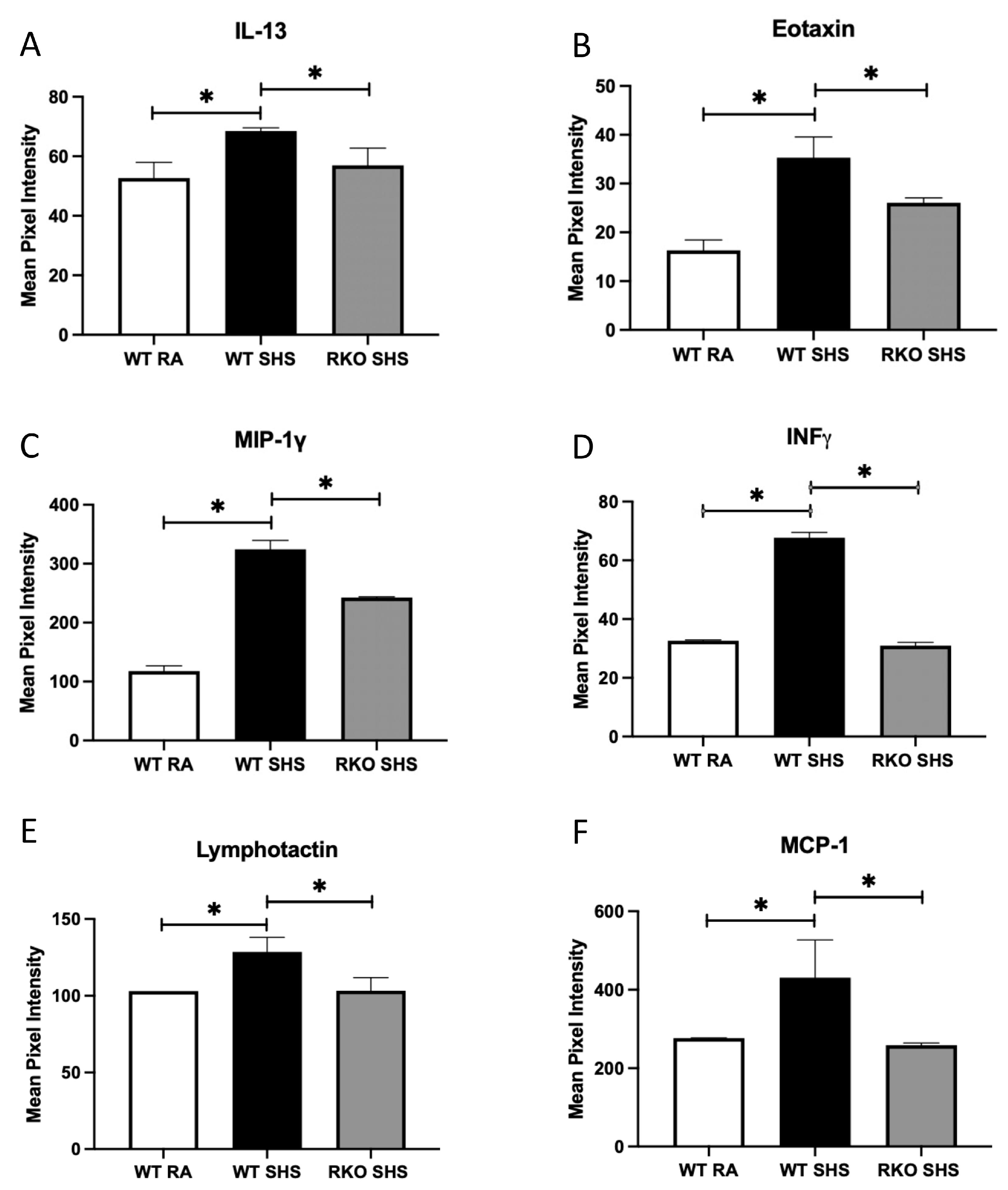
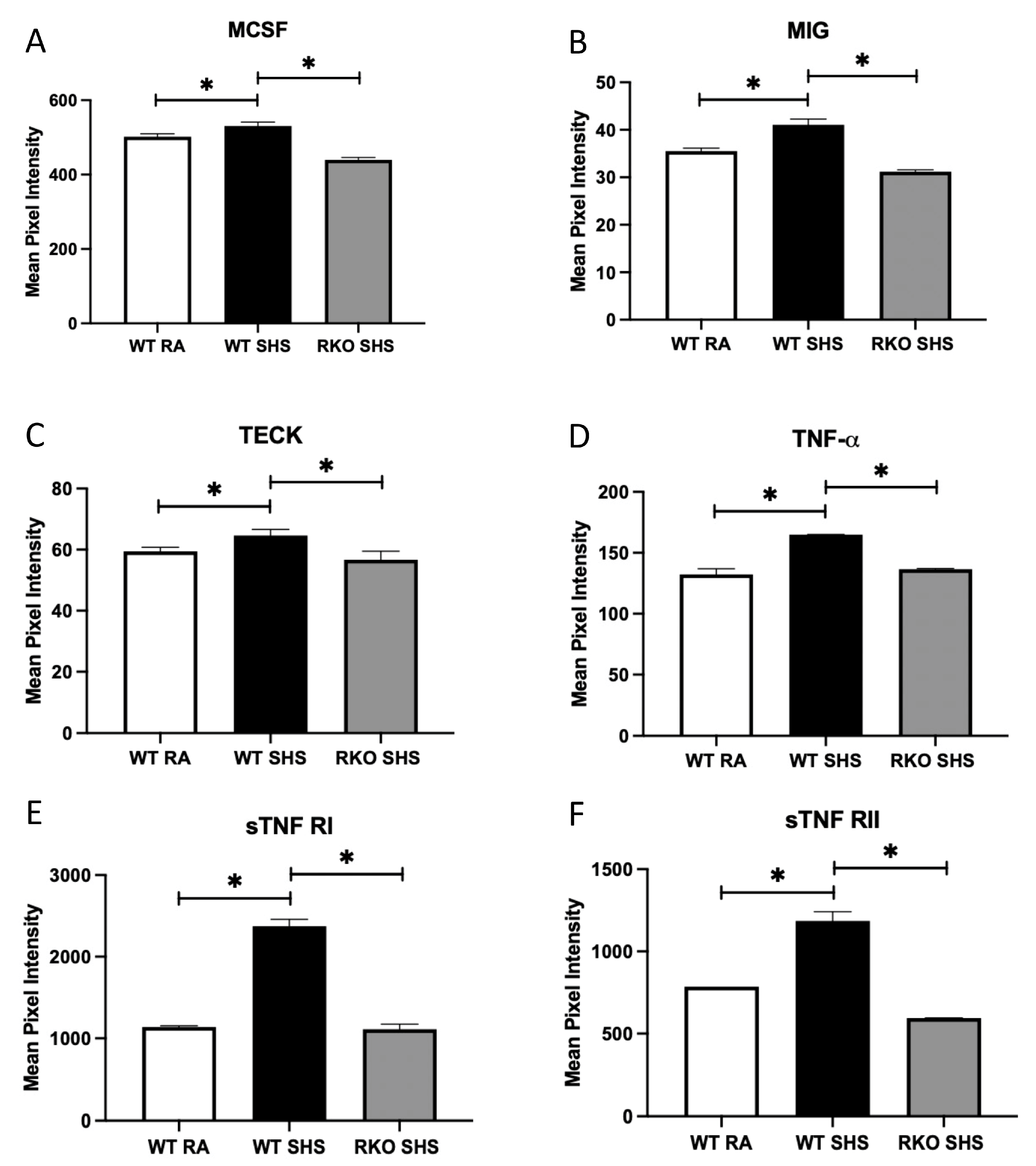
2.2. BALF Cellularity and Cytokine Abundance Following Chronic SHS Exposure
3. Discussion
4. Methods and Materials
4.1. Animals and Exposure to SHS
4.2. Ras and NF-kB Analyses
4.3. Immunoblot Analyses
4.4. Bronchoalveolar Lavage Fluid (BALF) Analyses
4.5. Inflammatory Molecule Analyses
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Staff, M.C. COPD. Available online: https://www.mayoclinic.org/diseases-conditions/copd/diagnosis-treatment/drc-20353685 (accessed on 12 September 2023).
- Eisner, M.D.; Anthonisen, N.; Coultas, D.; Kuenzli, N.; Perez-Padilla, R.; Postma, D.; Romieu, I.; Silverman, E.K.; Balmes, J.R. An Official American Thoracic Society Public Policy Statement: Novel Risk Factors and the Global Burden of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 182, 693–718. [Google Scholar] [CrossRef]
- MacNee, W. Pathology, pathogenesis, and pathophysiology. BMJ 2006, 332, 1202. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Raja, A.; Brown, B.D. Chronic Obstructive Pulmonary Disease; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- WHO. The Top 10 Causes of Death. World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 16 May 2023).
- Lamprecht, B.; McBurnie, M.A.; Vollmer, W.M.; Gudmundsson, G.; Welte, T.; Nizankowska-Mogilnicka, E.; Studnicka, M.; Bateman, E.; Anto, J.M.; Burney, P.; et al. COPD in Never Smokers. Chest 2011, 139, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.C.; Sin, D.D.; Bourbeau, J.; Hernandez, P.; Chapman, K.R.; Cowie, R.; FitzGerald, J.M.; Marciniuk, D.D.; Maltais, F.; Buist, A.S.; et al. Characteristics of COPD in never-smokers and ever-smokers in the general population: Results from the CanCOLD study. Thorax 2015, 70, 822–829. [Google Scholar] [CrossRef]
- Fu, Z.; Jiang, H.; Xu, Z.; Li, H.; Wu, N.; Yin, P. Objective secondhand smoke exposure in chronic obstructive pulmonary disease patients without active smoking: The U.S. National Health and Nutrition Examination Survey (NHANES) 2007–2012. Ann. Transl. Med. 2020, 8, 445. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.R.; Halbert, R.; Nordyke, R.J.; Schau, B. Airway obstruction in never smokers: Results from the Third National Health and Nutrition Examination Survey. Am. J. Med. 2005, 118, 1364–1372. [Google Scholar] [CrossRef]
- Eisner, M.D.; Balmes, J.; Yelin, E.H.; Katz, P.P.; Hammond, S.K.; Benowitz, N.; Blanc, P.D. Directly measured secondhand smoke exposure and COPD health outcomes. BMC Pulm. Med. 2006, 6, 12. [Google Scholar] [CrossRef]
- Yin, P.; Jiang, C.; Cheng, K.; Lam, T.; Lam, K.; Miller, M.; Zhang, W.; Thomas, G.; Adab, P. Passive smoking exposure and risk of COPD among adults in China: The Guangzhou Biobank Cohort Study. Lancet 2007, 370, 751–757. [Google Scholar] [CrossRef]
- Sezer, H.; Akkurt, I.; Guler, N.; Marakoğlu, K.; Berk, S. A Case-Control Study on the Effect of Exposure to Different Substances on the Development of COPD. Ann. Epidemiol. 2006, 16, 59–62. [Google Scholar] [CrossRef]
- Tsai, J.; Homa, D.M.; Gentzke, A.S.; Mahoney, M.; Sharapova, S.R.; Sosnoff, C.S.; Caron, K.T.; Wang, L.; Melstrom, P.C.; Trivers, K.F. Exposure to Secondhand Smoke Among Nonsmokers—United States, 1988–2014. Morb. Mortal. Wkly. Rep. 2018, 67, 1342. [Google Scholar] [CrossRef]
- Walton, K.; Gentzke, A.S.; Murphy-Hoefer, R.; Kenemer, B.; Neff, L.J. Exposure to Secondhand Smoke in Homes and Vehicles Among US Youths, United States, 2011–2019. Prev. Chronic Dis. 2020, 10, 17E103. [Google Scholar] [CrossRef]
- Diver, W.R.; Jacobs, E.J.; Gapstur, S.M. Secondhand Smoke Exposure in Childhood and Adulthood in Relation to Adult Mortality Among Never Smokers. Am. J. Prev. Med. 2018, 55, 345–352. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.; Brett, J.; Yan, S.; Wang, F.; Pan, Y.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.A. Role of the receptor for advanced glycation end products (RAGE) in inflammation. Investig. Clin. 2010, 51, 257–268. [Google Scholar]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Nicholl, I.D.; Bucala, R. Advanced glycation endproducts and cigarette smoking. Cell. Mol. Biol. 1998, 44, 1025–1033. [Google Scholar]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in lung disease: The receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as Mediator of NF-kB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar]
- Body-Malapel, M.; Djouina, M.; Waxin, C.; Langlois, A.; Gower-Rousseau, C.; Zerbib, P.; Schmidt, A.-M.; Desreumaux, P.; Boulanger, E.; Vignal, C. The RAGE signaling pathway is involved in intestinal inflammation and represents a promising therapeutic target for Inflammatory Bowel Diseases. Mucosal Immunol. 2019, 12, 468–478. [Google Scholar] [CrossRef]
- Del Turco, S.; Basta, G. An update on advanced glycation endproducts and atherosclerosis. BioFactors 2012, 38, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Vannucci, S.J.; Du Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-R.; Cho, W.-H.; Kim, J.; Lee, H.J.; Chung, C.; Jeon, W.K.; Han, J.-S. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp. Mol. Med. 2014, 46, e75. [Google Scholar] [CrossRef]
- Demling, N.; Ehrhardt, C.; Kasper, M.; Laue, M.; Knels, L.; Rieber, E.P. Promotion of cell adherence and spreading: A novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2005, 323, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.T.; Winden, D.R.; Marlor, D.R.; Wright, A.J.; Jones, C.M.; Chavarria, M.; Rogers, G.D.; Reynolds, P.R. Acute secondhand smoke-induced pulmonary inflammation is diminished in RAGE knockout mice. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L758–L764. [Google Scholar] [CrossRef]
- Hirschi-Budge, K.M.; Tsai, K.Y.F.; Curtis, K.L.; Davis, G.S.; Theurer, B.K.; Kruyer, A.M.M.; Homer, K.W.; Chang, A.; Van Ry, P.M.; Arroyo, J.A.; et al. RAGE signaling during tobacco smoke-induced lung inflammation and potential therapeutic utility of SAGEs. BMC Pulm. Med. 2022, 22, 160. [Google Scholar] [CrossRef]
- Reynolds, P.R.; Kasteler, S.D.; Schmitt, R.E.; Hoidal, J.R. Receptors for advanced glycation end-products signals through Ras during tobacco smoke-induced pulmonary inflammation. Am J Respir Cell Mol Biol. 2011, 45, 411–418. [Google Scholar] [CrossRef]
- Muller, W.A. Getting Leukocytes to the Site of Inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.-L.; Wautier, M.-P. Endothelial Cell Participation in Inflammatory Reaction. Int. J. Mol. Sci. 2021, 22, 6341. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Swigart, L.B.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Santarpia, M.; Aguilar, A.; Chaib, I.; Cardona, A.F.; Fancelli, S.; Laguia, F.; Bracht, J.W.P.; Cao, P.; Molina-Vila, M.A.; Karachaliou, N.; et al. Non-Small-Cell Lung Cancer Signaling Pathways, Metabolism, and PD-1/PD-L1 Antibodies. Cancers 2020, 12, 1475. [Google Scholar] [CrossRef]
- Liu, S.-S.; Liu, C.; Lv, X.-X.; Cui, B.; Yan, J.; Li, Y.-X.; Li, K.; Hua, F.; Zhang, X.-W.; Yu, J.-J.; et al. The chemokine CCL1 triggers an AMFR-SPRY1 pathway that promotes differentiation of lung fibroblasts into myofibroblasts and drives pulmonary fibrosis. Immunity 2021, 54, 2042–2056.e8. [Google Scholar] [CrossRef]
- De la Garza, M.M.; Cumpian, A.M.; Daliri, S.; Castro-Pando, S.; Umer, M.; Gong, L.; Khosravi, N.; Caetano, M.S.; Ramos-Castañeda, M.; Flores, A.G.; et al. COPD-Type lung inflammation promotes K-ras mutant lung cancer through epithelial HIF-1α mediated tumor angiogenesis and proliferation. Oncotarget 2018, 9, 32972–32983. [Google Scholar] [CrossRef]
- Sharma, A.; Kaur, S.; Sarkar, M.; Sarin, B.C.; Changotra, H. The AGE-RAGE Axis and RAGE Genetics in Chronic Obstructive Pulmonary Disease. Clin. Rev. Allergy Immunol. 2020, 60, 244–258. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Chao, L.; Chu, D.; Kalantar-Zadeh, K.; George, J.; Young, H.A.; Liu, G. Cytokines: From clinical significance to quantification. Adv Sci (Weinh) 2021, 8, e2004433. [Google Scholar] [CrossRef]
- Liu, D.; Xu, W.; Tang, Y.; Cao, J.; Chen, R.; Wu, D.; Chen, H.; Su, B.; Xu, J. Nebulization of risedronate alleviates airway obstruction and inflammation of chronic obstructive pulmonary diseases via suppressing prenylation-dependent RAS/ERK/NF-κB and RhoA/ROCK1/MLCP signaling. Respir. Res. 2022, 23, 380. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.; Asrat, S.; Allinne, J.; Lim, W.K.; Nagashima, K.; Birchard, D.; Srivatsan, S.; Ajithdoss, D.K.; Oyejide, A.; Ben, L.-H.; et al. IL-4 and IL-13, not eosinophils, drive type 2 airway inflammation, remodeling and lung function decline. Cytokine 2023, 162, 156091. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.G.; Rennick, D.; Donaldson, D.D.; Venkayya, R.; McArthur, C.; Hansell, E.; Kurup, V.P.; Warnock, M.; Grünig, G. IL-13 and IFN-γ: Interactions in Lung Inflammation. J. Immunol. 2001, 167, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Mason, R.J. The effect of interleukin-13 (IL-13) and interferon-γ (IFN-γ) on expression of surfactant proteins in adult human alveolar type II cells in vitro. Respir. Res. 2010, 11, 157. [Google Scholar] [CrossRef]
- Ito, Y.; Al Mubarak, R.; Roberts, N.; Correll, K.; Janssen, W.; Finigan, J.; Mishra, R.; Chu, H.W. IL-13 induces periostin and eotaxin expression in human primary alveolar epithelial cells: Comparison with paired airway epithelial cells. PLoS ONE 2018, 13, e0196256. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Cho, J.-H.; Cho, S.S.; Bae, C.-S.; Kim, G.-Y.; Park, D.-H. Establishment of a chronic obstructive pulmonary disease mouse model based on the elapsed time after LPS intranasal instillation. Lab. Anim. Res. 2018, 34, 1–10. [Google Scholar] [CrossRef]
- Kochetkova, E.A.; Nevzorova, V.A.; Ugai, L.G.; Maistrovskaia, Y.V.; Massard, G. The Role of Tumor Necrosis Factor Alpha and TNF Superfamily Members in Bone Damage in Patients with End-Stage Chronic Obstructive Lung Disease Prior to Lung Transplantation. Calcif. Tissue Int. 2016, 99, 578–587. [Google Scholar] [CrossRef]
- Goebeler, M.; Schnarr, B.; Toksoy, A.; Kunz, M.; Bröcker, E.; Duschl, A.; Gillitzer, R. Interleukin-13 selectively induces monocyte chemoattractant protein-1 synthesis and secretion by human endothelial cells. Involvement of IL-4R α and Stat6 phosphorylation. Immunology 1997, 91, 450–457. [Google Scholar] [CrossRef]
- Chen, M.; Wang, T.; Shen, Y.; Xu, D.; Li, X.; An, J.; Dong, J.; Li, D.; Wen, F.; Chen, L. Knockout of RAGE ameliorates mainstream cigarette smoke-induced airway inflammation in mice. Int. Immunopharmacol. 2017, 50, 230–235. [Google Scholar] [CrossRef]
- Amy, R.W.; Bowes, D.; Burri, P.H.; Haines, J.; Thurlbeck, W.M. Postnatal growth of the mouse lung. J. Anat. 1977, 124, 131–151. [Google Scholar]
- Sampath, V.; Davis, K.; Senft, A.P.; Richardson, T.R.; A Kitzmiller, J.; Berclaz, P.Y.; Korfhagen, T.R. Altered Postnatal Lung Development in C3H/HeJ Mice. Pediatr. Res. 2006, 60, 663–668. [Google Scholar] [CrossRef]
- ImageJ. Available online: https://imagej.nih.gov/ij/ (accessed on 13 March 2022).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curtis, K.L.; Homer, K.M.; Wendt, R.A.; Stapley, B.M.; Clark, E.T.; Harward, K.; Chang, A.; Clarke, D.M.; Arroyo, J.A.; Reynolds, P.R. Inflammatory Cytokine Elaboration Following Secondhand Smoke (SHS) Exposure Is Mediated in Part by RAGE Signaling. Int. J. Mol. Sci. 2023, 24, 15645. https://doi.org/10.3390/ijms242115645
Curtis KL, Homer KM, Wendt RA, Stapley BM, Clark ET, Harward K, Chang A, Clarke DM, Arroyo JA, Reynolds PR. Inflammatory Cytokine Elaboration Following Secondhand Smoke (SHS) Exposure Is Mediated in Part by RAGE Signaling. International Journal of Molecular Sciences. 2023; 24(21):15645. https://doi.org/10.3390/ijms242115645
Chicago/Turabian StyleCurtis, Katrina L., Kyle M. Homer, Ryan A. Wendt, Brendan M. Stapley, Evan T. Clark, Kaden Harward, Ashley Chang, Derek M. Clarke, Juan A. Arroyo, and Paul R. Reynolds. 2023. "Inflammatory Cytokine Elaboration Following Secondhand Smoke (SHS) Exposure Is Mediated in Part by RAGE Signaling" International Journal of Molecular Sciences 24, no. 21: 15645. https://doi.org/10.3390/ijms242115645
APA StyleCurtis, K. L., Homer, K. M., Wendt, R. A., Stapley, B. M., Clark, E. T., Harward, K., Chang, A., Clarke, D. M., Arroyo, J. A., & Reynolds, P. R. (2023). Inflammatory Cytokine Elaboration Following Secondhand Smoke (SHS) Exposure Is Mediated in Part by RAGE Signaling. International Journal of Molecular Sciences, 24(21), 15645. https://doi.org/10.3390/ijms242115645