Abstract
Chronic heart diseases, such as coronary heart disease, heart failure, secondary arterial hypertension, and dilated and hypertrophic cardiomyopathies, are widespread and have a fairly high incidence of mortality and disability. Most of these diseases are characterized by cardiac arrhythmias, conduction, and contractility disorders. Additionally, interruption of the electrical activity of the heart, the appearance of extensive ectopic foci, and heart failure are all symptoms of a number of severe hereditary diseases. The molecular mechanisms leading to the development of heart diseases are associated with impaired permeability and excitability of cell membranes and are mainly caused by the dysfunction of cardiac Ca2+ channels. Over the past 50 years, more than 100 varieties of ion channels have been found in the cardiovascular cells. The relationship between the activity of these channels and cardiac pathology, as well as the general cellular biological function, has been intensively studied on several cell types and experimental animal models in vivo and in situ. In this review, I discuss the origin of genetic Ca2+ channelopathies of L- and T-type voltage-gated calcium channels in humans and the role of the non-genetic dysfunctions of Ca2+ channels of various types: L-, R-, and T-type voltage-gated calcium channels, RyR2, including Ca2+ permeable nonselective cation hyperpolarization-activated cyclic nucleotide-gated (HCN), and transient receptor potential (TRP) channels, in the development of cardiac pathology in humans, as well as various aspects of promising experimental studies of the dysfunctions of these channels performed on animal models or in vitro.
1. Introduction
Calcium ions are vital for the normal functioning of the heart. They support the myocardial contraction of the heart and play a major role in regulating excitation–contraction coupling. Calcium current impacts the pacemaker activity and action potential formation (plateau phase) in cardiomyocytes. In addition, calcium ions are involved in cellular signaling, carrying out other general biological cellular functions, such as gene transcription, cell growth, and cell remodeling [1].
Cardiac Ca2+ channels are classified by their calcium selectivity and conductance as well as structure, the kinetics of activation and inactivation, and pharmacological characteristics [2].
The condition of these channels is regulated by (1) changes in the membrane potential effecting their gate mechanism (voltage-gated channels, VGCCs) [3]; (2) depletion of Ca2+ stores (store-operated Ca2+ channels) [4]; (3) Ca2+ or other intracellular stimuli (transient receptor potential (TRP) channels) [5]; (4) hyperpolarization and cyclic nucleotides (Ca2+-permeable nonselective cation HCN channels) [6]; and some others [7,8]. The predominant Ca2+ channels in the heart are highly selective voltage-gated channels, Cav channels (L- and T-types); however, an additional contribution to the provision of heart functions is made by nonselective ion channels that pass Ca2+ into cardiomyocytes (transient receptor potential (TRP) channels and hyperpolarization and cyclic nucleotide (HCN) channels [1,3,4,5,6,8].
Impairment of the physiological functions of Ca2+ channels leads to mitochondrial Ca2+ overload, damage to CM, apoptosis, and heart pathology. Currently, significant progress has been made in understanding the function of Ca2+ channels in a healthy heart, but there is still a limited amount of data on cytoarchitectonics and the physiology of Ca2+ channels in the cardiovascular cells associated with heart diseases. The dysfunction of voltage-gated calcium channels that occurs during pathophysiological processes in the heart leads to arrhythmias caused by early afterdepolarization (EAD) [9]. It has been shown in experimental animal models and in vitro that an overload of cardiomyocytes with calcium ions caused by the dysfunction of nonselective calcium channels resulted in delayed afterdepolarization (DAD) and irregular conduction cycles [10].
The problems of the origin and treatment of congenital and acquired Ca2+ channelopathies, the influence of Ca2+ channel mutations on the pathogenesis of chronic heart diseases, and associated multisystem disorders are also far from being fully understood. In addition, the contribution of posttranslational modifications of Ca2+ channels and the defects of Ca2+ handling proteins to the development of potentially lethal heart diseases are still poorly understood [11,12].
The purpose of this review is to highlight the little-studied aspects of the effect of Ca2+ channel dysfunction on the development of chronic and hereditary heart diseases.
2. Cardiac Voltage-Gated Calcium Channels (VGCCs)
The voltage-gated calcium channels (Cav channels) are an integral element of the sarcolemma of cardiomyocytes (CMs). Unlike other transport systems of cell membranes (carriers, pumps), they are able to pass Ca2+ at a rate several orders of magnitude higher in the open state. This determines their dominant role in ensuring the basic functions of the heart (contractility and pacemaking). The influx of Ca2+ contributes to keeping the membrane potential more positive, and the released calcium from the sarcoplasmic reticulum induces heart contraction [2]. This is a well-known phenomenon called “calcium-induced calcium release”. As a result, the excitation–contraction coupling process in cardiac myocytes can proceed without interruption [1].
Cardiomyocytes contain three types (L-, R-, and T-) of voltage-gated calcium channels that activate upon membrane depolarization. They differ in their properties, functions, and distribution in different compartments of the heart. Most importantly, these channels differ in the activation threshold, which is essential for ensuring the cellular specialization of cardiomyocytes [1,3,6,13,14].
2.1. L-Type Cav Channels (Cav1)
The main cardiac Cav channels are L-type Cav channels (LTCC—long-lasting large-capacity). These are high-threshold (HVA) ion channels, the activation threshold of which is significantly higher than in low-voltage-activated T-type calcium channels (LVA) [15]. It should be noted that Cav1 channels have electrophysiological properties that are characterized by high conductivity and very slow inactivation kinetics [2,15,16]. These properties of the channels ensure the generation of the action potential (in the plateau phase), the maintenance of normal sinus rhythm, and the excitation–contraction coupling (ECC) of cardiac cells in the depolarization phase of the action potential (AP) [17]. The L-type Cav channels (Cav1.2 and Cav1.3) are mainly localized in contractile cardiomyocytes and pacemaker cells of the cardiac conduction system [1,2,3,17,18]. It has been established that even minor changes in the function of calcium channels disrupt the plateau phase of the cardiac action potential and lead to an irregular conduction cycle [17], arrhythmias, and heart disease-associated pathophysiological conditions [1].
Cav1 channels are heterooligomeric protein complexes consisting of five subunits: α1, α2, δ, β, and γ. The α1 pore-forming subunit is the main component of the channel, and the remaining four subunits are auxiliary components (Figure 1).
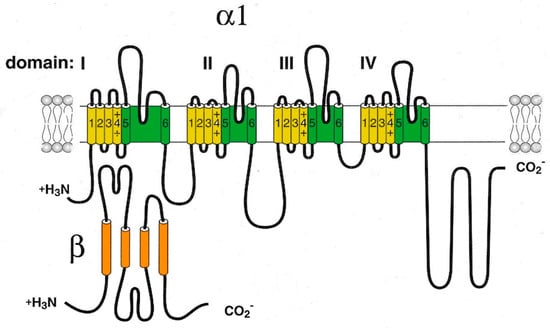
Figure 1.
Schematic representation of the pore-forming channel α1-subunit Cav1. The α1-subunit includes four homologous domains (I–IV), each of which consists of six transmembrane segments (1–6). Segments 5 and 6 together with the linker peptide (linkers 5–6) form a selective pore permeable to Ca2+ ions. Segment 4 is a voltage-sensing module. The β-subunit is involved in the inactivation and closure of the channel. Both N- and C-termini are in the cytosol.
However, the functioning of the channel and its positioning on the membrane requires the participation of all protein subunits [16]. It is noted that expression of the α1 subunit is sufficient to produce functional Ca2+ channels (in the example of skeletal muscles), but it has a low expression level and abnormal kinetics, and its voltage is dependent in the Ca2+ current [19]. Moreover, a few other effectors and regulatory proteins directly associated with α1-subunit are required to regulate Ca2+ transport and gating. These proteins (small and large GTPases, calmodulin, etc.) form supramolecular signal complexes with the α1-subunit of the Cav1 channel and significantly expand the repertoire of mechanisms that regulate the Ca2+ channel influx (Figure 2).
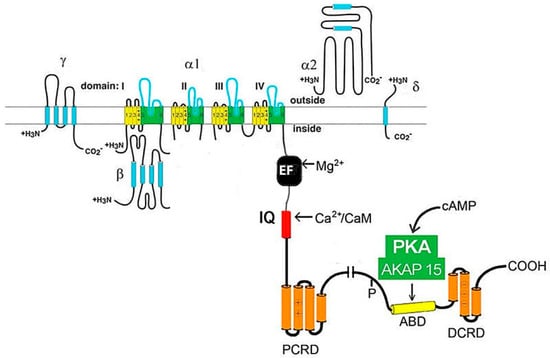
Figure 2.
The cardiac CaV1.2 channel signaling complex. ABD, AKAP15 binding domain; DCRD, distal C-terminal regulatory domain; PCRD, proximal C-terminal regulatory domain; scissors, site of proteolytic processing.
Previously, in animal models and in vitro, the mechanisms of the stimulating effect of catecholamines on myocardial contractility were studied, and it was found that the activation of β-adrenergic receptors increases L-type Ca2+ currents through PKA-mediated phosphorylation of the Cav1.2 channel protein and/or associated proteins [20]. To date, a lot of data have accumulated, indicating the important role of these signal complexes in physiological processes and cardiac pathophysiological conditions [1,3].
Voltage-gated calcium channels LTCC exist in three, Cav1.1, Ca1.2, and Cav1.3 (Cav1.1–1.3), isoforms, two of which, Cav1.2 and Cav1.3, are found in CM and slightly differ in the structure of α-subunits encoded by the CACNA1C and -D genes, respectively [16,21]. Cav 1.2 and Cav 1.3 channels possess high conductivity (25 pSm), very slow inactivation kinetics (τ > 500 ms), and the ability to activate at high membrane potentials (over −10 mV).
In adult cardiac myocytes, calcium flows through the Cav1.2 channel and forms the main type of internal current during the plateau phase of the cardiac AP, and Cav1.2 is the dominant channel involved in ECC. Calcium currents also influence the electrical properties of cardiomyocytes. Cardiomyocytes’ channel dysfunctions affecting the AP profile are associated with various cardiac arrhythmias. Studies conducted using a canine ventricular cell model have shown that the inactivation of Cav1 channels, including Ca-dependent inactivation, is associated with acute Ca2+/calmodulin-dependent protein kinase II (CaMKII) overexpression and calcium handling dysfunction [22].
Ventricular myocytes have only Cav1.2 channels, whereas both Cav1.2 and Cav1.3 channels are expressed in atrial myocytes, the Gis–Purkinje conducting system, SA and AV node pacemaker cells, and the smooth muscle cells of blood vessels. Nowadays, alternative LTCC splicing has attracted attention as an instrument of tissue specificity, which revealed that the dominant variant of the Cav1.2 channel in smooth muscle cells differs from the one in heart cells [2,21]. The activation threshold of Cav1.2 channels is less negative (−30 mV) than in Cav1.3 channels (−50 mV), which is essential to ensure the sequence of electrical activity in the myocardium [12,18]. Failure of the throughput function of Ca channels (increase or decrease in the time of their open state, even without a genetic change in their structure) can lead to serious impairment of the heart function. Under some conditions, the dysfunction of L-type calcium channels will cause early afterdepolarization, a common aspect of cardiac arrhythmias [9,10].
Under some conditions, the dysfunction of L-type calcium channels will cause early afterdepolarization (EAD), which arises on the shoulder of a preceding action potential plateau and is favored by a slow preceding activation rate and prolonged action potentials. In this case, Cav1.2 channels are responsible for the inward current for EADs. These afterdepolarizations have characteristics that suggest their etiological role in cardiac arrhythmias found in heart failure [23].
The role of Cav1.3 channels is not limited only to their participation in the generation of diastolic depolarization in pacemaker cells. The opening of these channels contributes to the generation of local diastolic intracellular Ca2+ releases (LCRs) and is required for the coupled-clock system that drives the automaticity of human sinoatrial nodal pacemaker cells [24]. Unsurprisingly, malfunction of these channels leads to SAN dysfunction, atrioventricular conduction disorders, arrhythmias, and heart failure [17,18].
Mutations found in gene-encoding Cav1 channels determine a wide range of diseases called calcium channelopathies, and all four gene-encoding α- and β-subunits carry such mutations [17]. Cav1-channelopathies include muscular, neurological, cardiac, and visual syndromes. Among them is Timothy syndrome, which is manifested by prolongation of the QT interval and congenital heart defects [25]. This condition is associated with a high risk of sudden cardiac death (SCD) and is caused by defects in the CACNA1C gene encoding in the α1-subunit of the Cav1.2 channel [25,26]. Mutations in the CACNA1C gene change the structure of α-subunits and the conformation of Cav1.2 channels. As a result of these mutations, these channels remain open longer than usual, which leads to an excessive intake of Ca2+ into the heart cells, an increase in cellular excitability, and an increased risk of life-threatening cardiac arrhythmias. The CACNA1C gene is located on the short arm of chromosome 12 (12p13.3) [26].
There are two molecular genetic variants of Timothy syndrome. The most common one is named the “classic variant”. It is caused by a mutation in exon 8a of the CACNA1C gene and is characterized by polymorphism of clinical manifestations associated with the expression of this gene site in various tissues of the body. The “atypical” variant is less common and caused by mutations in exon 8 of the CACNA1C gene, leading to more pronounced prolongation of the QT and QTc interval and ventricular arrhythmias, most of which are drug-induced or associated with the use of anesthesia [25]. The “atypical” variant of Timothy syndrome is characterized by the maximum expression of the CACNA1C gene in exon 8 in the heart and brain (80% CACNA1C mRNA) [25].
QT syndrome type V (SQT5) is another genetic heterogeneous disease associated with impaired functioning of Cav1.2 channels. It is characterized by a decrease in QT interval ≤ 300 ms and the appearance of a high symmetrical peak-shaped T wave. Mutations leading to a shortening of the AP, which are pathological for this syndrome, were found in the gene-encoding K+ channels and the CaCNB2b gene (locus 10p12.33) encoding the β2-subunit of Cav1.2 [27].
The CACNB2b gene encodes 660 amino acids of the β2-subunit Cav1.2 [28]. This gene is mainly expressed in heart cells. The influx of Ca2+ ions into the cytosol is reduced in the channels with defective β2-subunits, which leads to a decrease in the IL,Ca current. Phenotypically, mutations in the CACNB2b gene can lead to not only shortening of the QT interval (SQT5) but also to Brugada syndrome (BS) or a combination of both [28].
Brugada syndrome is known as a form of cardiac channelopathy associated with a high risk of SCD. It is believed that BS accounts for 20% of mortality in young people and men aged 30–40 years without structural pathology of the myocardium. The diagnostic hallmark of the syndrome is distinctive changes in the electrocardiogram (ECG) in the form of right bundle branch block and elevation of the ST segment in the right pericardial leads (V1-V3) [17,29]. This pathology is associated with mutations of the CACNA1C and CACNB2b gene-encoding α- and β2-subunits of the Cav1.2 channel, respectively. In patients with mutations of these genes, the bandwidth of Cav1.2 is reduced, and the ILCa current is decreased [27].
The role of Cav1.3 in rhythmogenesis and heart rate modulation has been established only recently [14]. Compared to other calcium channels, Cav1.3 channels are activated faster and at more hyperpolarized voltages, which is important in maintaining the pacemaking and regulation of heart automaticity [24].
In humans, the first channelopathy involving the CACNA1D gene-encoding Cav1.3 protein was identified in 2011 [14]. Initially, a loss-of-function mutation in an alternatively spliced exon was associated with congenital deafness. Further observations showed that patients with this pathology had bradycardia and SA node dysfunction (sinus node weakness syndrome) with preserved normal QRS and QT in ECG. In the experimental conditions on mice, the CACNA1D gene knockout was shown to lead to the appearance of viable and fertile offspring but with deafness, bradycardia, and the dysfunction of SA and AV nodes, resulting in pathological functional disorders close to their human counterparts [14]. Experimental data suggest that the C-terminus of the Cav1.3 protein can function as a transcription regulator in atrial CM and modulate the expression of the myosin II light chain and small conductance calcium-activated K+ channel [14].
In ventricular CM, L-type Cav channels are the main structures maintaining myocardial contractility. Genetic or posttranslational modifications of Cavα1- and Cavβ2-subunits can lead to significantly reduced left-ventricular contractility and the development of ventricular tachycardia [30]. Defects in these channels lead to an increase in the duration of their open state. As a result, the mechanism of Ca2+-induced Ca2+ release (CICR) from the sarcoplasmic reticulum (SR), which is the trigger for the onset of contraction, becomes disrupted. The L-type Cav channels’ activity in the ventricular CM are usually transient and beneficial, but chronic irritation can become pathological [31]. To restore the functions of the CM associated with the malfunction of Cav1.2 and Cav1.3 channels, a method of targeted mobilization of a pre-synthesized pool of subsarcolemmal Cav1.2 channel-containing vesicles/endosomes into the CM sarcolemma has been developed [31].
2.2. R-Type Ca2+ Channels (Cav2.3)
R-type Ca2+ channels (Cav2.3) are encoded by the CACNA1E gene and are intermediate in electrophysiological properties between L- and T-type Cav channels. These channels are found in the pacemaker cells of the brain and heart, where they provide repetitive firing of action potentials [32]. Among other representatives of calcium channels, pharmacoresistant R-type channels are considered the most “mysterious” [33]. Their structure has not been sufficiently studied. Most of them are encoded by the CACNA1E gene and are expressed as different Cav2.3 splice variants (variant Cav2.3a to Cav2.3e or Cav2.3f) as the ion conducting subunit [8]. In humans, an R-type channel has been found so far only in thalamic neurons but proposes a cardiac mechanism of action for this channel [34]. The basic information on these channels in cardiomyocytes was obtained in animal models and isolated cells [8,32,33,34,35,36]. Owning to research on experimental models, it is now clear that cardiac pharmacoresistant R-type Ca2+ channels play an important role in regulating heart rate, pacemaking, and cardiac conduction.
These channels are highly conserved, and it can be assumed that human homologs of R-channels have similar properties to animal channels. However, further studies are needed to perform a comparison of animal and human R-channel data.
Like other HVA Ca2+ channels, Cav2.3 channels form multi-subunit complexes consisting of pore-forming α1- and α2-subunits, one of several cytoplasmic β-subunits and extracellular δ-subunits. The Cavα1-subunit is a pseudotetrameric protein with four homologous repeats (I–IV), which consist of six spiral segments penetrating the membrane (S1–S6). Four segments (S1–S4) in each repetition form a voltage sensor module, while the remaining two segments (S5–S6) of all repeats make up the majority of the pore domain (PD) and activation gates. The pore and the selective filter possessing a conserved Ca2+-selectivity filter motif ([T/S] × [D/E] × W) are formed by re-enterable pore loops (p-loops) between segments S5 and S6, which partially re-enter the pore region. Three inter-domain linkers and phosphorylation sites in the cytoplasmic domain of the channel are involved in inactivation, the association of auxiliary Cavß-subunits, and intracellular modulation [8].
R-type Ca2+ currents have been known to be resistant to most subtype-specific organic and peptide Ca2+ channel blockers [37], but they are also blocked by Ni2+ and are sensitive to Zn2+ and pharmacological regulators of store-operated Ca2+ channels, in particular, to isoproterenol [8]. These data are important for further research in the field of studying the possible contribution of R-type Ca2+ channel dysfunctions to the development of chronic human heart diseases and the pursuit of promising blockers for their therapy.
So far, no inherited disease is known for the CACNA1E gene, but recently, spontaneous mutations leading to early death in humans were identified [34].
2.3. T-Type Ca2+ Channels
T-type Ca2+ channels (Cav3.1, Cav3.2) («transient», «short-term») (low threshold or fast transient) mean the opening time of the channel [2,3].
Unlike HVA, which opens at −20 mV, Cav3 channels have a low threshold since their activation happens near −40 mV (in SAN cells, the channel activation threshold is −55 mV) [18,21]. The Cav3 channels are characterized by high sensitivity to the blocking action of Ni2+, low sensitivity to dihydropyridines and amiloride, and low conductivity (~8 pSm). The activity of the channels is regulated by G-protein-coupled receptors (GPCRs), which are blocked by mibefradil, Ni2+ (especially Cav3.2), and curtoxin [3,19].
T-type calcium channels consist of a single pore-forming α1-subunit that has two key structural determinants of Cav channel gating as well as ion selectivity and permeability. The Cav3 pore-forming α1-subunit is a relatively large plasma membrane protein of about 260 kDa organized into four hydrophobic domains (DI–DIV), each of which consists of six transmembrane helices (S1–S6) (Figure 3).
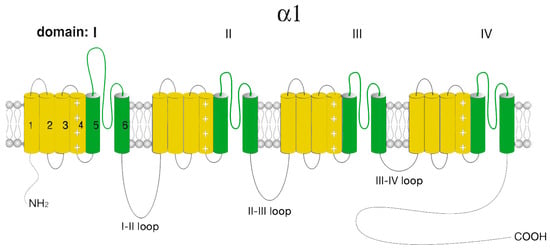
Figure 3.
Schematic representation of the Cav3 α1-subunit. The α1-subunit includes four homologous hydrophobic domains (I–IV), each of which consists of six transmembrane segments (1–6). Segments 5 and 6 together with linkers 5–6 form a highly conserved pore loop permeable to Ca2+ ions. Segment 4 is a voltage-sensing module. The amino (NH2 and carboxyl (COOH) termini and the cytoplasmic interdomain l-II-, II-III-, III-IV loops are in the cytosol.
Similar to L-type Ca2+ channels, the voltage-sensitive channel module (S4) is formed by positively charged rich arginine/lysine, while the selectivity and channel ion conductivity depend on the re-entrant linkers connecting S5 and S6 modules and forming a P-loop. The four TM modules are linked together by several intracellular loops connecting the S6 module of the upstream domain to the S1 module of the downstream domain, which in combination with the NH2- and COOH-termini, provide a site/center for channel regulation by various signaling molecules and other protein partners, including the βγ-dimer of the G-proteins, PKA, calcineurin, CaMKII, syntaxin−1A, stac1, CACHD1, spectrin α/β, ankyrin B, etc. Furthermore, T-type channels undergo several posttranslational modifications, such as phosphorylation, glycosylation, and ubiquitination, which contribute to the expression and activity of the channel [3,38].
T-type calcium channels exhibit variations in their electrophysiological and pharmacological properties that can be explained by the existence of several channel splice variants [39]. These variants include Cav3.1, Cav3.2, and Cav3.3, which are encoded by the genes CACNA1G, CACNA1H, and CACNA1I, respectively, in humans [21,39].
T-type channels are formed by numerous variants of α1-subunits, among which the Cav3.1 channels made of (α1G) subunits are found in the heart. These channels are expressed mainly in sinoatrial and atrioventricular node cells, where along with Cav1.3 channels (α1D), they play an important role in generating spontaneous excitation of pacemaker cells. T-type channelopathies that drastically impair cardiac automaticity are considered rare [40]. They are associated with severe hereditary diseases that lead to sudden cardiac death. One such disease is myotonic dystrophy type I (MD1), known as Steinert disease, associated with a DMPK gene defect and channelopathy caused by mutations in genes associated with cardiac function (i.e., TNNT, TNNT2, TTN, TPM1, SYNE1, MTMR1, NEBL, and TPM1), including CACNA1A and CACNA1H [41].
Patients with MD1 suffer from defects in conductivity and atrial or ventricular tachyarrhythmia. The disease progresses with aging and becomes complicated by second- and third-degree heart blockage and left ventricular hypertrophy [41]. Histopathological analysis of the affected hearts in patients with MD1 showed fibrosis and multifocal disintegration of myofibrils [41].
Cav3.1 channel dysfunction was detected in patients with sinus node weakness and heart blockage caused by congenital autoimmune disease of the cardiac conduction system [24].
In ventricular CM, the population of Cav3.1 and Cav3.2 channels is uncommon. Therefore, their role in the regulation of myocardial contractility is insignificant. Transient expression of T-type Ca2+ channels occurs in the embryonic heart [42]. In a mouse model, it was demonstrated that the Cav3.2 channels are predominantly expressed in the embryonic heart from 9.5 to 18 days of embryonic development. At the same time, Cav3.1 channels are also expressed, but their expression level is significantly lower than Cav3.2 [43]. The functional role of these channels in the embryonic heart remained unknown. The T-type Ca2+ channels were suggested to be involved not only in the regulation of cell proliferation of prenatal CM but may also be included in the processes of cell growth in the differentiated heart [43]. Indeed, it turned out that Cav3.1 and Cav3.2 channels can be re-expressed in ventricular myocytes with pathological hypertrophy and myocardial infarction [43,44]. However, the increased expression of Cav3.2 channels is limited to the myocardial lesion zone and has a regional and temporary nature [43]. The hypertrophic overloads of the heart from α1G-transgenic mice demonstrated that the mice were resistant to pressure overload, isoproterenol, and cardiac hypertrophy caused by physical exertion. These mice also had no cardiac pathology, despite a significant increase in the influx of Ca2+ into CM. Unlikely, α1G-/-mice showed enhanced hypertrophic reactions after cardiac overload by pressure or the infusion of isoproterenol. Pathological hypertrophy in α1G-/-transgenic mice was reversed using the α1G-transgene, which proves the importance of Cav3.1 channels in cardioprotection and the prevention of cell remodeling [43].
3. Store-Operated Calcium Ca2+ Channels
Ca2+ ions play an important role in many physiological processes, including pacemaking, contraction, the release of neurotransmitters, ECC, gene expression, etc. Unsurprisingly, with a relatively low [Ca2+]i, a significant amount of Ca2+ is preserved by cells in intracellular Ca2+ stores. In CM and vascular smooth muscle cells (VSMCs), Ca2+ is stored mainly in SR. Calcium levels in cells are precisely regulated by various transporters and ion channels. Reduction in the intra-organelle concentration of Ca2+ serves as a signal to refill the Ca2+ stores through a store-operated calcium entry mechanism (SOCE).
3.1. Cardiac Ryanodine Receptor (RyR2)
Ryanodine receptors (RyR2s) are Ca2+-permeable ion channels in the membrane of the SR. These channels are responsible for local Ca2+-induced Ca2+ release from the SR. The Ca2+ released (Ca2+ sparks) activates contraction, pacemaking, and ECC, and affects other Ca2+-dependent intracellular processes.
RyRs are found in both atrial and ventricular CMs, as well as in vascular smooth muscle cells [45].
Currently, three isoforms of the RyRs have been characterized, one of which is the type 2 ryanodine receptor (RyR2), which has been found in CM. Modern innovations in cryo-electron microscopy have made it possible to obtain a number of near-atomic RyR structures that have contributed to a better understanding of the RyR architecture [46]. The RyR2 consists of four subunits combined into a homotetramer and the FC-binding protein calstabin 2 in a stoichiometric ratio of 1:4 [47].
The distinctive feature of the channels formed by RyR2 is that they are activated by an extracellular influx of Ca2+ as a result of the Cav1-RyR2 interaction that triggers local Ca2+ release [46].
The structure of these channels, as well as the molecular mechanisms responsible for cardiac pathology induced by the dysfunction of RyR2 and other cardiac Ca channels, were considered in detail in fundamental review papers published earlier [48,49]. The RyR2 macromolecular complex is one of the most well-studied multiprotein complexes [49]. A diverse array of RyR2-interacting proteins directly regulates RyR2 channel activity by binding to the pore subunit (e.g., FK506-binding protein−12.6 (FKBP12.6), calmodulin (CaM), calsequestrin-2 (CASQ2), junctin (JCTN), triadin (TRDN), and βII-spectrin [49]. CASQ2 binds to RyR2 via both JCTN and TRDN. RyR2 is strongly regulated by luminal Ca2+ levels, either by direct Ca2+ binding to RyR2 or by luminal Ca2+ interacting with CASQ2, JCTN, and TRDN [50]. Other proteins in the RyR2 macromolecular complex regulate the level of RyR2 posttranslational modification [49].
RyR2s are part of the pacemaker molecular mechanism that ensures heart automaticity. The contribution of the RyR2-dependent Ca2+ releases (Ca2+ sparks) to the automaticity is currently interpreted within the framework of a model dubbed the “calcium clock” [51]. According to the coupled-clock pacemaker cell system concept, the “clock” in pacemaker cells forms two competing oscillators: the “Ca2+-clock” mechanism based on the spontaneous release of Ca2+ from the SR and the “membrane clock” (“M-clock”), which includes surface membrane cation channels to ignite an AP [51]. The phenomenon of Ca2+ oscillations, underlying the “Ca2+-clock”, is associated with the local diastolic intracellular Ca2+ releases (LCRs), and it is independent of the membrane potential (MP). Indeed, spontaneous LCRs can be observed in the absence of changes in the MP and are a distinctive feature of the “Ca2+-clock” [52]. RyRs act as a gear in a “Ca2+-clock”, inducing rhythmical discharges of LCRs, which, in turn, activate an inward current (INCX1) that prompts the “M-clock” to start an AP [51]. This electrogenic transport mechanism generates an internal ion current, which contributes to the onset of diastolic depolarization. In the final phase of diastolic depolarization, Cav channels cooperate with a Na+/Ca2+-exchanger (NCX) to raise the MP to the threshold value of the AP [51]. During the AP phase, the intake of Ca2+ through LTCC channels refills the leakage of Ca2+ from the SR, allowing new diastolic intracellular Ca2+ release to occur in the next cycle. Consequently, under normal physiological conditions, the “Ca2+-clock” and the “M-clock” synchronize the pacemaker activity of the heart cells and create a reliable basis for heart automaticity [51,53].
Considering the importance of the RYR2 in providing heart automaticity, it seems quite obvious that mutations of genes encoding these receptors or proteins interacting with RYR2 in the CICR would lead to catastrophic consequences for the organism [54]. Indeed, studies using a mouse model have shown that a RYR2 mutation with a locus in the CaM-binding site (reducing receptor inactivation) causes cardiac hypertrophy, heart failure, and early sudden death [55,56].
Clinical studies suggest that enhancing the interaction of CaM-RyR2 may represent an effective therapeutic strategy for the treatment of cardiac arrhythmias and heart failure. In support of this idea, the mutation of GOF CaM-M37Q and the reinforcement of the CaM-RyR2 interaction have been demonstrated to be able to suppress the spontaneous release of Ca2+ from the SR and catecholaminergic polymorphic ventricular tachycardia [57].
In chronic heart diseases accompanied by cardiac arrhythmias, there is an increase in the activity of Cav1.2 channels, which leads to an increase in their permeability to Ca2+ and, as a result, to CM calcium overload. At the same time, spontaneous Ca2+ releases induced by more frequent RYR2 openings form “pathological” calcium waves that are abolished and removed under physiological conditions with the participation of NCX1 and other molecular determinants of Ca2+ homeostasis (CASQ2, FKBP12, SERCA2a, etc.) [46]. It was found that NCX1 dysfunction and changes in its expression profile during arrhythmia lead to changes in atrial cell morphology and calcium handling together with dramatic alterations in the function of SAN [58,59].
It should be noted that the occurrence of pathological Ca2+ waves or an increase in the “threshold” of waves may be caused not only by the dysfunction of the cardiac calcium channels, in particular RyR2, but also by other pathogenetic factors, such as hypoxia and acidosis. Ca2+ alternans in ischemia can be taken as the arrhythmic triggers, leading to afterdepolarization and the substrate facilitating re-entry by inducing electrical alternans. More information about the cellular mechanism of cardiac alternans is found in the other studies [60].
Recent studies have shown that spontaneous arrhythmogenic “calcium waves” can result from genetic mutations of RyR2 but are more often due to an increase in the time of its open state [61,62,63,64,65]. Defects in its modular coupling with regulatory proteins of the cytosol, such as CaM, Epacl, PDE, FKBP12.6, PKA, PP1, calstabin, etc., or with Ca2+-binding proteins localized in the lumen of the SR (junctin, triadin, and calsequestrin), form temporary macromolecular complexes and can lead to a disruption of the RyR2 gate function [62,66,67,68,69]. Mechanisms of interaction of the partner molecules with RyR2 are built on a structural basis, while regulatory proteins (predominantly kinases) use RyR2 as a scaffold protein to form functional signal complexes that can modify a large number of other Ca2+-dependent molecules involved in the cascade signal transmission. The structure of these RyR2 multi-domain complexes and the mechanisms of regulation of their activity are still far from being fully understood [63,69].
It is noted that point mutations of RyR2-associated proteins or changes in their expression can dramatically affect the development of cardiac arrhythmias [70]. In particular, the expression level of serine/threonine protein phosphatases plays an important role in the pathogenesis of arrhythmias [71]. Serine/threonine protein phosphatases (PP1, PP2A, and PP2B) control the dephosphorylation of numerous cardiac proteins, including a variety of ion channels (Cav1.2, NKA, NCX, etc.), calcium-handling proteins (SERCA, junctin, and PLB), contractile proteins, MLC2, TnI, and MyBP-C [71,72], thereby providing posttranslational regulation of ECC and other heart functions. Accordingly, the dysfunction of this regulation can contribute to the development of cardiac arrhythmias. Atrial fibrillation (AF) is the most common heart rhythm disorder, and it is characterized by electrical and structural cardiac remodeling that among other factors includes changes in the phosphorylation status of a wide range of proteins, such as RyR2 [71]. It was found that a decrease in the concentration of PP1 caused by an increase in the level of PP1 regulatory proteins like inhibitor-1 (I-1), inhibitor-2, or heat-shock protein 20 in the sarcoplasm of ventricular CM leads to the rapid development of tachycardia and could cause sudden death [63,72], although an experimental increase in the concentration of PP1 in sarcoplasm prevented the development of arrhythmia, which was proven in experiments on mice overexpressing Ang II [73]. Mice characterized with a highly phosphorylated RyR2-S2808 site (S2808A+/+) demonstrated an increased sensitivity of RyR2 to Ca2+ during the dephosphorylation of PP-1 [74].
CaM kinase II is another enzyme that plays an important pathogenetic role in diseases accompanied by cardiac arrhythmia [52,54,75]. The effect of CaM kinase II on RyR2 is controversial. In pharmacological experiments using the method of embedding proteins in an artificial bilayer, some authors revealed the activating effect of this kinase on RyR2. On the other hand, others demonstrated its inhibitory effect [76]. Using molecular genetic methods (transgenic overexpression), more recent studies have found that CaM kinase II binds RyR2 and causes its phosphorylation at serine 2814 (RYR2-S2814). This, in turn, increases the frequency of Ca2+ spikes and the spontaneous local diastolic subsarcolemmal Ca2+ releases in the process of ECC [52]. It was noted that an increase in the level of phosphorylation of RYR-S2814 in SAN pacemaker cells led to the alteration of the “Ca2+-clock” regulation and the development of heart failure [77].
The inhibition of CaM kinase II by a specific blocker KN93 reduced the release of Ca2+ from the SR and slowed the heart rate [52]. In vitro studies have shown the possibility of using this blocker to relieve ventricular tachycardia caused by oxidative stress, which opens up prospects for the therapeutic use of KN-93 and its functional analogs in the treatment of arrhythmias [52].
In addition, an increase in the basal level of CaMKII through the phosphorylation of histone deacetylases (HDACs) activates myocyte-enhancer factor 2 (MEF2), which initiates the CaMKII/MEF-2 signaling pathway and hypertrophic remodeling of ventricular myocytes [78]. Among endogenous biologically active molecules that have arrhythmogenic effects, the most studied are neurohormones, such as endothelin-1 (ET-1), epinephrine, norepinephrine, etc., which act through Gαq-associated GPCRs and cAMP-dependent protein kinase A (PKA) [79,80,81]. Increased hormonal stimulation of these receptors leads to the hyperphosphorylation of RyR2 and the dissociation of calstabin 2 (FKBP12.6) from it. RyR2, deprived of this protein, loses its locking function, which leads to an increase in the time of its open state, a leakage of Ca2+, an increase in [Ca2+]i, and afterdepolarization, which can cause “fatal arrhythmia”, heart attack, and SCD [82]. To date, two genetic diseases associated with mutations in ventricular RyR2 have been described: catecholaminergic polymorphic ventricular tachycardia (CPVT), or familial polymorphic ventricular tachycardia, and arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) type 2 [83]. In patients with CPVT, the affinity of calstabin 2 to RyR2 is reduced due to a defect in RyR2 at its binding site to calstabin [82]. The use of molecular approaches in the strategy of the targeted therapy of CVD resulted in new drugs that suppress the hyperphosphorylation of p-RyR2 (Ser2808) and p-RyR2 (Ser2814), thereby stabilizing RyR2 and normalizing heart rate and the contractility of ventricular myocytes [84]. The ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 19 October 2023) describes 165 variants of pathogenic mutations of the RYR2 gene. The most common mutations associated with CPVT are Ser2246Leu, Arg2474Ser, Asn4104Lys, Arg4497Cys, Pro2328Ser, 1.1-KB DEL, and EX3 [85]. Most of these mutations lead to amino acid substitution or the appearance of a premature stop codon and a disruption of the formation of a functional protein [86].
In addition to the RYR2 gene, the encoding ryanodine receptor calcium release channel and mutations in five gene-encoding proteins from the SR calcium-release complex are involved in the pathogenesis of CPVT: CASQ2 (encoding cardiac calsequestrin), TRDN (encoding triadin), CALM1, CALM2, and CALM3 (encoding identical protein calmodulin) [87].
The development of arrhythmogenic dysplasia of the right ventricle is also associated with mutations in the RYR2 gene. The ARVD2 locus was mapped to chromosome 1q42–q43 [88]. This disease is characterized by partial fatty or fibrous degeneration of the myocardium of the right ventricle, electrical instability, and sudden death [88]. The detection of RyR2 mutations causing CPVT and ARVD2 opens the way to the pre-symptomatic detection of carriers of the disease in childhood, enabling early monitoring and treatment [87,88].
3.2. Ion Channels with Transient Receptor Potential (TRPC, TRPM7, TRPA1)
Transient receptor potential channels are nonselective cation channels of the TRP channel superfamily, uniting the related receptor proteins capable of being activated by the potential originating from the binding of the ligand to the receptor [89]. This superfamily is divided into a family of canonical TRP channels (TRPC) and several families whose names come from the name of the receptor, binding to which initiates the potential. Most TRPs are polymodal channels, so-called «coincidence detectors» that are activated by both physical and chemical stimuli [89]. TRP channels vary in degrees of selectivity and permeability to ions. TRPV1–TRPA1 and TRPM6/7 channels are more selective for Ca2+ ions [90]. In CM, they are localized in the sarcolemma adjacent mainly to intercalated disks and are activated by phospholipase C (PLC) via Gaq-associated G-protein-coupled receptors [89].
The common structural features of these channels are four N-terminal ankyrin repeats, six short TM domains, and a pore-forming region localized between transmembrane domains 5 and 6. Like the other previously described channels, TRP channels with partner proteins and kinases form signaling complexes that can be involved in the pathogenesis of various cardiovascular diseases [91,92,93,94].
The existing data suggest that several types of TRP channels (TRPC3, TRPC6, TRPV1, TRPV3, TRPV4, TRPA1, TRPM6, and TRPM7) may play a central role in the progression of fibroproliferative disorders in the heart and blood vessels and contribute to both acute and chronic inflammatory processes involved in them [91,95].
The family of canonical TRP channels consists of proteins closely related to the Drosophila channel proteins of the same name involved in photoreception [96]. This family includes seven subfamilies (TRPC1–TRPC7), of which proteins of the TRPC1, -3, -4, -6, and -7 subfamilies were found in CM [97]. To date, the greatest interest is focused on mechanosensitive TRPC channels. In case of their molecular «breakdown», these channels begin to pass an increased flow of Ca2+ ions, and, thereby, activate processes involving the pathological remodeling of CM [91,97,98]. In addition, TRPC7 also mediates apoptosis, thereby contributing to the process of heart failure [99].
All TRPC channels are dependent on receptors associated with PLC since they are directly or indirectly activated by phospholipid products formed due to activation of this enzyme and the induction of the hydrolysis of membrane phospholipids. TRPV1, -2, and -5 channels are activated by binding IP3 to the receptors and are responsible for SOCE. In this case, the interaction of TRPC with Orai protein and stromal interacting molecule 1 (STIM1) is noted [100]. Coupling between the Ca2+ entry and the intracellular Ca2+ stored in CM and VSMCs is mainly mediated by stromal interaction molecule, STIM1, located in the endoplasmic reticulum and ORAI1 membrane protein. These proteins are the primary components of the calcium-release activated calcium (CRAC) channel (Figure 4) [100]. In response to a decrease in the concentration of Ca2+ in SR, STIM1 is homooligomerized and translocated to the SR-PM contact sites, where it colocalizes with Ca2+-ATPase (SERCA), IP3R, and Orai1, forming the Ca2+ selective Orai1 pore. However, it is still unclear how such a close arrangement of proteins is conducted [100].
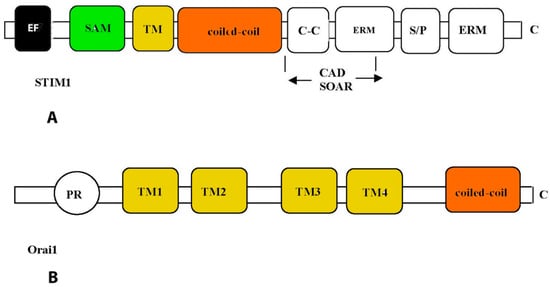
Figure 4.
Structural features of STIM1 (A) and Orai1 (B) proteins. Functional domains of proteins are enclosed in rectangles: EF—Ca2+-binding motif “EF-hands”; SAM—so-called “sterile”-α-motif (sterile-α-motif); TM—transmembrane domain; ERM—protein binding domain of the ERM complex; S/P—domain enriched with serine and proline; C-C—domain enriched with lysine; CAD—domain responsible for channel activation; SOAR, STIM1-Orai—activation region; PR—domain enriched with proline and arginine; TM1–4—transmembrane domains.
The mechanism of Ca2+ store filling with the participation of these proteins is as follows. After Ca2+ store depletion, the STIM1 protein located in the ER undergoes a complex conformational rearrangement, which results in STIM1 translocation into discrete ER plasma membrane junctions, where it directly interacts with the plasma membrane protein Orai1. Orai 1 triggers the recruitment of TRPC1 into the plasma membrane where it is activated by STIM1. TRPC1 and Orai1 form discrete STIM1-gated channels for the entry of Ca2+ into the lumen of the ER [101,102]. In addition, STIM1 can also activate TRPC1 through its C-terminal polybasic domain and is distinct from its Orai1-activating domain, SOAR [101].
TRPM2 is the second member of the TRPM subfamily that includes eight members, specifically TRPM1–8. TRPM2 is widely expressed in CM, where it forms a Ca2+-permeable cation channel and serves as a cellular sensor for oxidative stress or inflammatory response [103,104,105].
The N-terminus is composed of four melastatin homology regions, homology region pre-S1 (melastatin homology regions (MHRs), and homology regions (HRs). The channel domain contains six TMs (S1–S6), corresponding to a voltage-sensor-like domain; the pore is formed by the loop between S5 and S6. The C-terminus is composed of TRP and the coiled-coil domain (CC) (Figure 5).
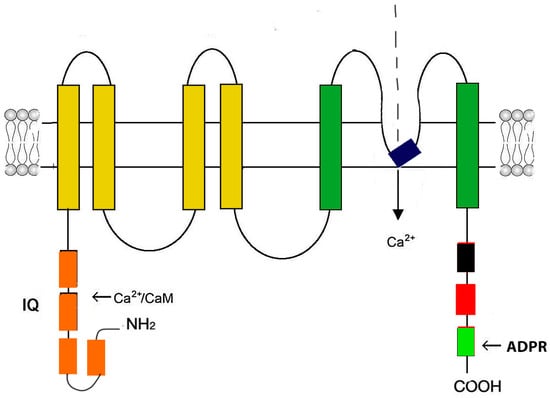
Figure 5.
Schematic representation of the Trpm2 monomer structure. The Trpm2 monomer consists of six transmembrane domains (yellow and dark green rectangles). The pore-firming loop is located between segments 5 and 6 (black rectangle). Both N- and C-termini are in the cytosol. The N-terminus contains four modules of the Trpm subfamily melastatin homology domain (MHD) (orange rectangles). In the second MHD, there is an IQ-like motif that binds Ca2+-calmodulin. The C-terminus contains a Trp box (TRP) (black rectangle), a coiled-coil domain (CC) (red rectangle), and a adenosine diphosphate ribose (ADPR) pyrophosphatase homolog domain.
TRPM2 channels are activated by ADPR, Ca2+, H2O2, and other reactive oxygen species (ROS). They serve as cellular sensors for oxidative stress, mediating oxidative stress-induced [Ca2+]i increase and contributing to pathological processes in many cell types, including CM. The overexpression of Trpm2 induces cell injury and death by Ca2+ overload or enhanced inflammatory response [103,105].
Mutations in genes encoding closely related TRPM4 channels lead to impaired automatism, conduction, and the appearance of hereditary progressive familial heart block type I (PFHBI) [106,107]. It is also assumed that some forms of provoked cardiac arrhythmia may occur due to a single gain-of-function mutation in TRPM4. To date, 47 mutations of the TRPM4 channel have been registered in the Human Gene Mutation Database [108,109].
TRPM7 channel mutations are especially dangerous in the prenatal period, as they can lead to intrauterine fatal arrhythmia and fetal death or a change in the myocardial transcription profile in adulthood and the deterioration of ventricular contractile function, conduction, and repolarization [110,111].
There is evidence of a wide involvement of TRP channels in the pathogenesis of CVD caused by hypoxia and oxidative stress, as well as ischemia-reperfusion (I/R) [90,104,112,113,114].
It is known that during hypoxia and I/R, there is a formation of ROS and an accumulation of lipid peroxidation products, including unsaturated aldehydes, such as acrolein and 4-hydroxynonenal, which are TRPA1 agonists, in cardiac tissue [115]. The mechanism of the toxic action of unsaturated aldehydes on CM is associated with their high electrophilicity and the ability to covalently bind to cysteine residues in the TRPA1 molecule, leading to the opening of these channels and an increase in sarcoplasmic reticulum Ca2+ release flux into the cytosol [116]. The overexpression of TRPC1 channels also contributes to Ca2+ leakage from the SR [117]. As a result, the Ca2+ overload of CM leads to impaired contractility, heart failure, and myocardial infarction [117].
The pathogenic significance of TRP channels in the development of heart failure, coronary artery disease, arterial and pulmonary hypertension, coronary microvascular dysfunction, and atherosclerosis is evident [91,93,94,118,119,120,121,122].
TRPA1, TRPV1–4, and TRPC1–6 are expressed on the surface of endothelial cells and ensure the passage of Ca2+ ions into cells, regulating endothelial-dependent vasodilation in response to a number of signaling molecules, such as endothelial-derived hyperpolarizing factor (EDHF), NO, and prostacyclin [90,113,119,123]. In mice with TRPV1 and TRPC3 channel knockout, there was a decrease in aortic vasodilation in response to carbachol, which proves the involvement of these channels in endothelium-dependent vasodilation [119,124]. In experiments on three animal models (dog, rat, and mouse), it was demonstrated that the i.v. administration of the TRPV4 agonist, GSK1016790A, stimulated endothelial-derived EDHF-dependent vasodilation and led to a subsequent decrease in blood pressure [125]. The TRPV4 channels have been noted to participate in the coupling of endothelial-dependent vasodilation and the relaxation of VSMCs through interaction with RyR2 and the big conduction BKCa channel [126]. TRPV4/7, TRPC1/5/6/7, and TRPV1/2/4 are expressed in VSMCs and participate in myogenic regulation of the vascular tone [119,120,127]. Electrophysiological experiments demonstrated that the TRPM4 knockout mice had an increased vascular tone and developed hypertension [90,128], while the inhibition of TRPC6 reduced VSMC contraction [127]. TRPV1 channels are involved in the progression of the atherosclerotic process in the apolipoprotein E gene knockout mice [129]. Further studies have shown that other mechanosensitive TRP channels may also play a role in the development of atherosclerosis and coronary heart disease [130,131]. In addition, a disruption of the TRP channel expression or function may explain the observed increased cardiovascular risk in patients with metabolic syndrome [132].
Thus, TRP channels have broad cardiovascular plasticity. TRPC3, TRPC5, TRPC6, TRPV1, and TRPM7 are involved in the vasoconstriction and regulation of blood pressure and can be considered potential therapeutic targets for the treatment of chronic CVD, including cardiometabolic diseases, and myocardial atrophy [91,119,121,132,133,134].
4. Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channels
HCN channels are nonselective cationic channels that are involved in the generation of pacemaker activity in heart and brain cells [135,136]. They belong to the family of channels operated by cyclic nucleotides, which are part of the superfamily of potential-operated potassium channels, the regulation of which is under the control of the autonomic nervous system [137]. There are four isoforms of HCN channels (HCN1–4) encoded by the same genes [138]. The function of HCN channels is to generate a the “funny current” (If) or “pacemaker current” (If) during the hyperpolarization phase of the AP [139,140].
HCN channels are expressed differentially. HCN4 is the dominant form found in the SAN. HCN2 is found in the His–Purkinje system. HCN1 is also expressed in the SAN, but it is less optimal for pacemaker targeting [139]. HCN1 and HCN2 transcripts are predominantly found in ventricular CM [140,141].
HCN channels are primarily selective to Na+ and K+, but their functioning directly affects the influx of Ca2+ ions into CM and has a regulatory effect on diastolic depolarization under physiological conditions [142,143,144,145]. Electrophysiological experiments using the patch-clamp technique revealed that the influx of Ca2+ through HCN2 channels is enhanced by increasing the time of their open state with an increase in the concentration of cAMP and is inhibited by the specific If-blocker ivabradine [145].
HCN channels are tetrameric cation channels [139,146]. They consist of four subunits (Figure 6), which can be either the same or different from each other [139]. However, in vivo channels consisting of subunits of the same type are more common [146].
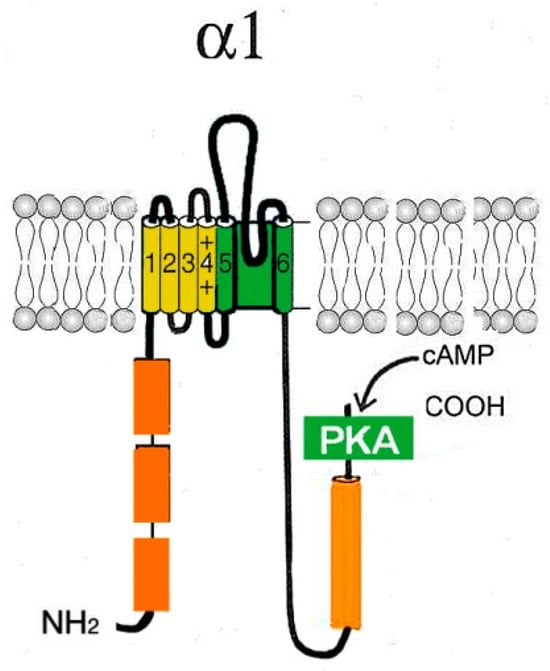
Figure 6.
Schematic representation of the α1-subunit of the HCN channel. Each HCN monomer contains six TM segments (1–6), including a positively charged potential sensor (S4) and P-region located between the pore-forming segments 5 and 6. The C-terminus contains two conserved structured regions: the C-linker, contributing to tetramerization, and the CNBD, which allows for modulation by cAMP. A region at the end of the N-terminal domain (orange rectangles) is conserved among HCN channel subtypes and has been found to be important for channel trafficking.
Additionally, HCN1 contains a di-arginine ER retention signal in the intrinsically disordered region of the C-terminus of HCN1 [147]. This signal controls the trafficking of HCN1 and negatively regulates the surface expression of HCN1 [146,147]. Deletion of the entire N terminus (residues 1–185) also prevented surface expression of HCN2 [148].
HCN isoforms (1–4) are highly conserved relative to TM and the binding site for cyclic nucleotides (80–90% identity) but have differences in activation and reactivation kinetics, depending on voltage and cAMP modulation [149,150]. For example, HCN4 exhibits the slowest activation and reactivation kinetics and opens at more negative potentials than other isoforms [151,152]. On the contrary, HCN1 demonstrates the fastest kinetics and opens at more positive potentials [152]. HCN4 is the most sensitive to cAMP, while the HCN1 subtype is faintly affected by cAMP and other cyclic nucleotides [139,153,154].
Cardiac activity is under hormonal control and modulated by mechanisms mediated by small proteins, β-adrenoreceptors, and cAMP [155,156]. An uncontrolled increase in the concentration of cAMP (sympathicotonia), or other non-canonical cyclic nucleotides, an increase in membrane expression, and the activation rate of HCN channels in the SAN can lead to modulation of the cAMP-dependent activation profiles and an increase in heart rate and SCD [155,157].
HCN channel dysfunction, decreased levels of connexin 40, connexin 43, and myocyte-enhancer factor-2C, and components of gap junction were noted in the hearts of transgenic mice with an overexpression of calreticulin [158]. The complex of these disorders underlies the development of arrhythmia, dilated cardiomyopathy, and SCD [158]. Age-related conduction disorders are also associated with HCN channel dysfunction [159].
Recent studies have shown the existence of four HCN channelopathies associated with different types of arrhythmia [153]. The mutation in exon 5 of the HCN4 gene is functionally associated with a truncated protein that is unable to bind cAMP and, therefore, has a dominant negative effect on channel function. Missense mutations of the same gene have been found in families of patients with SAN dysfunction. The presence of these mutations led to recurrent syncopes, severe bradycardia (39 beats per minute), prolonged QT intervals, and polymorphic ventricular tachycardia [153]. The dysfunction of HCN channels, mainly HCN4, is associated with sick sinus syndrome and other arrhythmias, such as atrial fibrillation, ventricular tachycardia, and atrioventricular block. In recent years, several data have also shown that dysfunctional HCN channels (HCN1, HCN2, and HCN4) may play an important role in the pathogenesis of epilepsy [160]. Myocardial involvement is frequent in patients affected by neuromuscular disorders and is the main cause of death in some conditions (Table 1).

Table 1.
Ca2+ channels and associated channelopathies (based on OMIM, the Online Mendelian Inheritance in Man database).
Currently, there are ongoing studies on the directed transport of recombinant genes directly into the heart [141]. In experiments on mice, adenoviral constructs expressing the HCN2 gene were delivered by epicardial injection into the root of the appendage of the left atrial appendage. Four days after the targeted delivery of the HCN2 gene, spontaneous beats occurring at the injection site were detected. The heart rate in experimental mice was under the control of the autonomic nervous system, which was proved by stimulation of the heart rate with catecholamines and a decrease in heart rate because of stimulation of the left vagus nerve. Cells localized at the injection site showed an increased expression of HCN2 channels and increased If currents [161].
These studies show the effectiveness of a targeted approach in the treatment of arrhythmias and congenital cardiac HCN channelopathies in humans [141,160].
However, for the successful gene therapy of HCN channelopathies in humans, additional mutant or chimeric HCN channel constructs will be required, which will have more positive activation and increased reactivity and will optimize the method of delivery of the therapeutic genes to the target cells of the heart. Research in this direction is underway [141,160].
5. Prospects of Using the Achievements of Molecular Biology in the Treatment of Chronic Heart Diseases
The cornerstone of the strategy of targeted molecular therapy is the idea of using drugs that act on subcellular structures involved in the mechanism of disease development. Currently, such drugs are mainly used for the treatment of certain types of cancer. In the therapy of cardiac diseases, the use of substances targeting certain molecular substrates has not yet become common. The most well-known drugs that can be attributed to this drug group are calcium channel blockers (CCBs). They have been used since the 1970s and have proven to be effective and reliable means in the treatment of CVD accompanied by rhythm disorders and decreased myocardial contractility. The broad use of CCBs in clinical practice was facilitated by their high anti-ischemic and antianginal efficacy and good tolerability, which were established during large clinical studies [162]. Over the past years, more than one generation of drugs of this group has changed. Modern CCBs (amlodipine, lacidipine, etc.) are substances that differ from their predecessors (verapamil, nifedipine, diltiazem, etc.) by the prolongation of action and a higher safety profile [163,164]. Drugs of this group have a cardioprotective effect by improving myocardial perfusion, reducing myocardial oxygen demand, and reducing the formation of free radicals and mitochondrial Ca2+ overloading of CM [163,164,165]. The disadvantages of CCBs as drugs are their rather wide range of action and low selectivity with respect to their molecular targets directly in the myocardium. Moreover, the use of these drugs is associated with an increased risk of developing proarrhythmia and systemic toxicity, an increase in the defibrillation threshold, and, in some cases, an increase in mortality [166]. Such significant disadvantages of CCBs make it necessary to search for novel alternative drugs, the targets of therapeutic action of which are not the channel proteins themselves but the molecules modulating their activity. Such proteins can be fully attributed to CaM kinase II, which is, on the one hand, a key modulating enzyme of Ca2+ metabolism in chronic heart pathology, and on the other hand, a widespread protein found in other vertebrates, making it possible to study the effect of CaM kinase II blockers initially in vivo experiments and in situ [167,168].
It is obvious that other regulatory proteins that exert their cardiotropic effect through interaction with Ca2+ channels or their modulators can become targets for pharmacological strategy in the treatment of chronic heart diseases. We mentioned some of them previously [84,85]. Currently, the most promising are studies aimed at finding blockers of the NCX1 [169], various isoforms of phosphodiesterases [170], and subunits of voltage-operated channels of various types, including nonselective HCN1–4 and TRP channels [171,172]. The use of these “molecular tools” to influence the mechanisms of Ca2+ signaling in heart failure and other chronic heart diseases is the first step toward making new drugs whose therapeutic targets are small fragments of specialized molecules or their specific isoforms. Recent progress in the studies of the structure and properties of Ca2+ channels and Ca2+ handling proteins, as well as modern technologies using targeted nanoparticles and targeted gene delivery directly to the heart, allow us to hope that significant progress will be made in the treatment of severe CVD associated with heart rate and contractility disorders in the near future [141,173].
6. Conclusions
Most chronic heart diseases are characterized by disorders of heart rhythm, conduction, and contractility. In addition, a violation of the electrical activity of the heart, the appearance of extensive ectopic foci, and heart failure are all symptoms of a number of severe hereditary diseases. The molecular mechanisms leading to the development of heart diseases are associated with impaired permeability and excitability of cell membranes and are mainly caused by the dysfunction of cardiac Ca2+ channels. More than 100 varieties of ion channels have been found in the cells of the cardiovascular system. The relationship between the activity of these channels and cardiac pathology, as well as the general biological function of cardiac muscle cells, is intensively studied in experimental animal models in situ and in vivo. However, little is still known about the origin of genetic Ca2+ channelopathies in humans, the role of the dysfunctions of various types of Ca2+ channels in the phenomenon of cardiac alternans, and the development of cardiac pathology in humans. Most research in this area is conducted using genetically modified animal models or in vitro. To what extent are the data obtained correct in relation to people? This question is still difficult to answer. Some Ca2+ channels, for example, subpopulations of R-type Ca2+ channels, are found only in animals. Other channels (subpopulations of T-channels) arise in early ontogenesis and undergo involution during natal (perinatal) development. Do they matter in the pathophysiology of the heart? The role of nonselective (hyperpolarization-activated cyclic nucleotide-dependent channels (HCN) and transient receptor potential (TRP)) in the development of cardiac pathology requires clarification. There are still no answers to questions related to the specific treatment of Ca2+ channelopathies in humans. However, with the use of the latest innovative instrumental and molecular genetic research methods, new opportunities for solving these problems are emerging.
Funding
The work is supported by the IEPhB Research Program 075-00967-23-00.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| ADPR | ADP ribose |
| AP | action potential |
| ARVC/D | arrhythmogenic right ventricular cardiomyopathy/dysplasia type 2 |
| AVN | atrioventricular node |
| BS | Brugada syndrome |
| Ca2+ | calcium |
| [Ca2+]i | intracellular Ca2+ concentration |
| CAD | coronary artery disease |
| CaM | calmodulin |
| Cav channels | voltage-gated calcium channels |
| Cav1 | L-type Cav channels |
| CCBs | calcium channel blockers |
| CCs | calcium channelopathies |
| CHD | coronary heart disease |
| CIRC | Ca2+-induced Ca2+ release |
| CM | cardiomyocyte |
| CNBD | cyclic nucleotide-binding domain |
| CPVT | catecholaminergic polymorphic ventricular tachycardia |
| CRAC | calcium-release activated calcium channel |
| CVDs | cardiovascular diseases |
| DADs | afterdepolarizations |
| DM1 | myotonic dystrophy type I |
| ECC | excitation–contraction coupling |
| ECG | electrocardiogram |
| EDHF | endothelial-derived hyperpolarizing factor |
| GPCR | G-protein-coupled receptors |
| HF | heart failure |
| HR | heart rate |
| HVA | high-voltage-activated voltage-gated calcium channels |
| IHD | ischemic heart disease |
| I/R | ischemia/reperfusion |
| LCRs | local diastolic intracellular Ca2+ release |
| LTCC | L-type Cav channels (Cav1) |
| MI | myocardial infarction |
| MP | membrane potential |
| NCX1 | Na+/Ca2+ exchange |
| PDE | phosphodiesterase |
| PFHBI | progressive familial heart block type I |
| PKA | cAMP-dependent protein kinase A |
| PLC | phospholipase C |
| PM | plasma membrane |
| PP | resting potential |
| ROS | reactive oxygen species |
| RP | resting potential |
| RyR2 | ryanodine receptor type 2 |
| SAN | sinus node |
| SOCE | store-operated calcium entry |
| SQTS | short QT syndrome |
| SR | sarcoplasmic reticulum |
| STIM1 | stromal interacting molecule 1 |
| TM | transmembrane domain |
| TS | Timothy syndrome |
| TRP | transient receptor potential (canonical, vallinoid-related, melastatin-related) |
| SCD | sudden cardiac death |
| SMCs | smooth muscle cells |
| VGCC | voltage-gate calcium channel |
| VSMCs | vascular smooth muscle cells |
References
- Shah, K.; Seeley, S.; Schulz, C.; Fisher, J.; Gururaja Rao, S. Calcium Channels in the Heart: Disease States and Drugs. Cells 2022, 11, 943. [Google Scholar] [CrossRef]
- Bkaily, G.; Jacques, D. Calcium Homeostasis, Transporters, and Blockers in Health and Diseases of the Cardiovascular System. Int. J. Mol. Sci. 2023, 24, 8803. [Google Scholar] [CrossRef]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef]
- Collins, H.E.; Zhang, D.; Chatham, J.C. STIM and Orai Mediated Regulation of Calcium Signaling in Age-Related Diseases. Front. Aging 2022, 3, 876785. [Google Scholar] [CrossRef]
- Venkatachalamo, K.; Montell, C. TRP channels. Annu Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Depuydt, A.S.; Peigneur, S.; Tytga, J. Review: HCN Channels in the Heart. Curr. Cardiol. Rev. 2022, 18, e040222200836. [Google Scholar]
- Weisbrod, D. Small and Intermediate Calcium Activated Potassium Channels in the Heart: Role and Strategies in the Treatment of Cardiovascular Diseases. Front. Physiol. 2020, 11, 590534. [Google Scholar] [CrossRef]
- Schneider, T.; Neumaier, F.; Hescheler, J.; Alpdogan, S. Cav2.3 R-type calcium channels: From its discovery to pathogenic de novo CACNA1E variants: A historical perspective. Pflug. Arch. 2020, 472, 811–816. [Google Scholar] [CrossRef]
- Dai, L.; Zang, Y.; Zheng, D.; Xia, L.; Gong, Y. Role of CaMKII and PKA in Early Afterdepolarization of Human Ventricular Myocardium Cell: A Computational Model Study. Comput. Math. Methods Med. 2016, 2016, 4576313. [Google Scholar] [CrossRef]
- Fozzard, H.A. Afterdepolarizations and triggered activity. Basic Res. Cardiol. 1992, 87, 105–113. [Google Scholar] [CrossRef]
- Martin, C.A.; Huang, C.L.; Matthews, G.D. The role of ion channelopathies in sudden cardiac death: Implications for clinical practice. Ann. Med. 2013, 45, 364–374. [Google Scholar] [CrossRef]
- Striessnig, J. Voltage-Gated Ca2+/Channel α1-Subunit de novo Missense Mutations: Gain or Loss of Function–Implications for Potential Therapies. Front. Synaptic Neurosci. 2021, 13, 634760. [Google Scholar] [CrossRef]
- Galetin, T.; Tevoufouet, E.E.; Sandmeyer, J.; Matthes, J.; Nguemo, F.; Hescheler, J.; Weiergräber, M.; Schneider, T. Pharmacoresistant Cav2·3 (E-type/R-type) voltage-gated calcium channels influence heart rate dynamics and may contribute to cardiac impulse conduction. Cell Biochem. Funct. 2013, 31, 434–449. [Google Scholar] [CrossRef]
- Priest, B.T.; McDermott, J.S. Cardiac ion channels. Channels 2015, 9, 52–359. [Google Scholar] [CrossRef]
- Bean, B.P. Two kinds of calcium channels in canine atrial cells. Differences in kinetics, selectivity, and pharmacology. J. Gen. Physiol. 1985, 86, 1–30. [Google Scholar] [CrossRef]
- Catterall, W.A. Signaling complexes of voltage-gated sodium and calcium channels. Neurosci. Lett. 2010, 486, 107–116. [Google Scholar] [CrossRef]
- Betzenhauser, M.J.; Pitt, G.S.; Antzelevitch, C. Calcium Channel Mutations in Cardiac Arrhythmia Syndromes. Curr. Mol. Pharmacol. 2015, 8, 133–142. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Couette, B.; Marger, L.; Bourinet, E.; Striessnig, J.; Nargeot, J. Voltage-dependent calcium channels and cardiac pacemaker activity: From ionic currents to genes. Prog. Biophys. Mol. Biol. 2006, 90, 38–63. [Google Scholar] [CrossRef]
- Nargeot, J.; Mangoni, M.E. Ionic channels underlying cardiac automaticity: New insights from genetically-modified mouse strains. Arch. Mal. Coeur Vaiss. 2006, 99, 856–861. [Google Scholar]
- Tsien, R.W. Adrenaline-like effects of intracellular iontophoresis of cyclic AMP in cardiac Purkinje fibres. Nat New Biol. 1973, 245, 120–122. [Google Scholar] [CrossRef]
- Baudot, M.; Louradour, J.; Torrente, A.G.; Fossier, L.; Talssi, L.; Nargeot, J.; Barrère-Lemaire, S.; Mesirca, P.; Mangoni, M.E. Concomitant genetic ablation of L-type Cav1.3 (α1D) and T-type Cav3.1 (α1G) Ca2+ channels disrupt heart automaticity. Sci. Rep. 2020, 10, 18906. [Google Scholar] [CrossRef]
- Zang, Y.; Dai, L.; Zhan, H.; Dou, J.; Xia, L.; Zhang, H. Theoretical investigation of the mechanism of heart failure using a canine ventricular cell model: Especially the role of up-regulated CaMKII and SR Ca(2+) leak. J. Mol. Cell. Cardiol. 2013, 56, 34–43. [Google Scholar] [CrossRef]
- Ducceschi, V.; Di Micco, G.; Sarubbi, B.; Russo, B.; Santangelo, L.; Iacono, A. Ionic mechanisms of ischemia-related ventricular arrhythmias. Clin. Cardiol. 1996, 19, 325–331. [Google Scholar] [CrossRef]
- Torrente, A.G.; Mesirca, P.; Bidaud, I.; Mangoni, M.E. Channelopathies of voltage-gated L-type Cav1.3/α1D and T-type Cav3.1/α1G Ca2+ channels in dysfunction of heart automaticity. Pflug. Arch. 2020, 472, 817–830. [Google Scholar] [CrossRef]
- Gakenheimer-Smith, L.; Meyers, L.; Lundahl, D.; Menon, S.C.; Bunch, T.J.; Sawyer, B.L.; Tristani-Firouzi, M.; Etheridge, S.P. Expanding the phenotype of CACNA1C mutation disorders. Mol. Genet. Genomic. Med. 2021, 9, e1673. [Google Scholar] [CrossRef]
- Harrell, D.T.; Ashihara, T.; Ishikawa, T.; Tominaga, I.; Mazzanti, A.; Takahashi, K.; Oginosawa, Y.; Abe, H.; Maemura, K.; Sumitomo, N.; et al. Genotype-dependent differences in age of manifestation and arrhythmia complications in short QT syndrome. Int. J. Cardiol. 2015, 190, 393–402. [Google Scholar] [CrossRef]
- Zhong, R.; Zhang, F.; Yang, Z.; Li, Y.; Xu, Q.; Lan, H.; Cyganek, L.; El-Battrawy, I.; Zhou, X.; Akin, I.; et al. Epigenetic mechanism of L-type calcium channel β-subunit downregulation in short QT human induced pluripotent stem cell-derived cardiomyocytes with CACNB2 mutation. Europace 2022, 24, 2028–2036. [Google Scholar] [CrossRef]
- Alings, M.; Wilde, A. “Brugada” syndrome: Clinical data and suggested pathophysiological mechanism. Circulation 1999, 99, 666–673. [Google Scholar] [CrossRef]
- Ednie, A.R.; Bennett, E.S. Intracellular O-linked glycosylation directly regulates cardiomyocyte L-type Ca2+ channel activity and excitation-contraction coupling. Basic Res. Cardiol. 2020, 115, 59. [Google Scholar] [CrossRef] [PubMed]
- Angelini, M.; Pezhouman, A.; Savalli, N.; Chang, M.G.; Steccanella, F.; Scranton, K.; Calmettes, G.; Ottolia, M.; Pantazis, A.; Karagueuzian, H.S.; et al. Suppression of ventricular arrhythmias by targeting late L-type Ca2+ current. J. Gen. Physiol. 2021, 153, e202012584. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.W.; Hannigan, K.I.; Ghosh, D.; Xu, B.; Del Villar, S.G.; Xiang, Y.K.; Dickson, E.J.; Navedo, M.F.; Dixon, R.E. β-adrenergic-mediated dynamic augmentation of sarcolemmal CaV1.2 clustering and co-operativity in ventricular myocytes. J. Physiol. 2019, 597, 2139–2162. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Dibue-Adjei, M.; Neumaier, F.; Akhtar, I.; Hescheler, J.; Kamp, M.A.; Tevoufouet, E.E. R-Type Voltage-Gated Ca2⁺ Channels in Cardiac and Neuronal Rhythmogenesis. Curr. Mol. Pharmacol. 2015, 8, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Neumaier, F.; Schneider, T.; Albanna, W. Cav2.3 channel function and Zn2+-induced modulation: Potential mechanisms and (patho)physiological relevance. Channels 2020, 14, 362–379. [Google Scholar] [CrossRef]
- Tevoufouet, E.E.; Nembo, E.N.; Dibué-Adjei, M.; Hescheler, J.; Nguemo, F.; Schneider, T. Cardiac functions of voltage-gated Ca(2+) channels: Role of the pharmacoresistant type (E-/R-Type) in cardiac modulation and putative implication in sudden unexpected death in epilepsy (SUDEP). Rev. Physiol. Biochem. Pharmacol. 2014, 167, 115–139. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Alpdogan, S.; Hescheler, J.; Neumaier, F. In vitro and in vivo phosphorylation of the Cav2.3 voltage-gated R-type calcium channel. Channels 2018, 12, 326–334. [Google Scholar] [CrossRef]
- Yao, X.; Wang, Y.; Wang, Z.; Fan, X.; Wu, D.; Huang, J.; Mueller, A.; Gao, S.; Hu, M.; Robinson, C.V.; et al. Structures of the R-type human Cav2.3 channel reveal conformational crosstalk of the intracellular segments. Nat. Commun. 2022, 13, 7358. [Google Scholar] [CrossRef]
- Randall, A.; Tsien, R.W. Pharmacological dissection of multiple types of calcium channel currents in rat cerebellar granule neurons. J. Neurosci. 1995, 15, 2995–3012. [Google Scholar] [CrossRef]
- Weiss, N.; Zamponi, G.W. Genetic T-type calcium channelopathies. J. Med. Genet. 2020, 57, 1–10. [Google Scholar] [CrossRef]
- Cribbs, L.L.; Lee, J.H.; Yang, J.; Satin, J.; Zhang, Y.; Daud, A.; Barclay, J.; Williamson, M.P.; Fox, M.; Rees, M.; et al. Cloning and characterization of alpha1H from human heart, a member of the T-type Ca2+ channel gene family. Circ. Res. 1998, 83, 103–109. [Google Scholar] [CrossRef]
- Vassort, G.; Talavera, K.; Alvarez, J.L. Role of T-type Ca2+ channels in the heart. Cell Calcium 2006, 40, 205–220. [Google Scholar] [CrossRef]
- Petri, H.; Vissing, J.; Witting, N.; Bundgaard, H.; Køber, L. Cardiac manifestations of myotonic dystrophy type 1. Int. J. Cardiol. 2012, 160, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Niwa, N.; Takemura, H.; Opthof, T.; Muto, T.; Horiba, M.; Shimizu, A.; Lee, J.K.; Honjo, H.; Kamiya, K.; et al. Pathophysiological significance of T-type Ca2+ channels: Expression of T-type Ca2+ channels in fetal and diseased heart. J. Pharmacol. Sci. 2005, 99, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Bodi, I.; Correll, R.N.; Chen, X.; Lorenz, J.; Houser, S.R.; Robbins, J.; Schwartz, A.; Molkentin, J.D. α1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice. J. Clin. Investig. 2009, 119, 3787–3796. [Google Scholar] [CrossRef]
- Min, J.Y.; Sandmann, S.; Meissner, A.; Unger, T.; Simon, R. Differential effects of mibefradil, verapamil, and amlodipine on myocardial function and intracellular Ca(2+) handling in rats with chronic myocardial infarction. J. Pharmacol. Exp. Ther. 1999, 291, 1038–1044. [Google Scholar]
- Kushnir, A.; Wajsberg, B.; Marks, A.R. Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Kurebayashi, N.; Yamazawa, T.; Murayama, T. Regulatory mechanisms of ryanodine receptor/Ca2+ release channel revealed by recent advancements in structural studies. J. Muscle Res. Cell Motil. 2021, 42, 291–304. [Google Scholar] [CrossRef]
- Hamilton, S.L.; Serysheva, I.I. Ryanodine receptor structure: Progress and challenges. J. Biol. Chem. 2009, 284, 4047–4051. [Google Scholar] [CrossRef]
- Correll, R.N.; Goonasekera, S.A.; van Berlo, J.H.; Burr, A.R.; Accornero, F.; Zhang, H.; Makarewich, C.A.; York, A.J.; Sargent, M.A.; Chen, X.; et al. STIM1 elevation in the heart results in aberrant Ca2+ handling and cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 87, 38–47. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Chen, H.; Valle, G.; Furlan, S.; Nani, A.; Gyorke, S.; Fill, M.; Volpe, P. Mechanism of calsequestrin regulation of single cardiac ryanodine receptor in normal and pathological conditions. J. Gen. Physiol. 2013, 142, 127–136. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Vinogradova, T.M.; Maltsev, V.A. The missing link in the mystery of normal automaticity of cardiac pacemaker cells. Ann. N. Y. Acad. Sci. 2008, 1123, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.B.; Val-Blasco, A.; Davoodi, M.; Gómez, S.; Yaniv, Y.; Benitah, J.P.; Gómez, A.M. Heart failure in mice induces a dysfunction of the sinus node associated with reduced CaMKII signaling. J. Gen. Physiol. 2022, 154, e202112895. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Mesirca, P.; Marqués-Sulé, E.; Zahradnikova, A., Jr.; Villejoubert, O.; D’Ocon, P.; Ruiz, C.; Domingo, D.; Zorio, E.; Mangoni, M.E.; et al. RyR2R420Q catecholaminergic polymorphic ventricular tachycardia mutation induces bradycardia by disturbing the coupled clock pacemaker mechanism. JCI Insight 2017, 2, e91872. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yao, J.; Belke, D.; Guo, W.; Zhong, X.; Sun, B.; Wang, R.; Paul Estillore, J.; Vallmitjana, A.; Benitez, R.; et al. Ca2+-CaM Dependent Inactivation of RyR2 Underlies Ca2+ Alternans in Intact Heart. Circ. Res. 2021, 128, e63–e83. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, T.; Oda, T.; Chakraborty, A.; Chen, L.; Uchinoumi, H.; Knowlton, A.A.; Fruen, B.R.; Cornea, R.L.; Meissner, G.; et al. Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ. Res. 2014, 114, 295–306. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Takahashi, N.; Xu, L.; Smithies, O.; Meissner, G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca2+ release channel. J. Clin. Investig. 2007, 117, 1344–1353. [Google Scholar] [CrossRef]
- Liu, B.; Walton, S.D.; Ho, H.T.; Belevych, A.E.; Tikunova, S.B.; Bonilla, I.; Shettigar, V.; Knollmann, B.C.; Priori, S.G.; Volpe, P.; et al. Gene Transfer of Engineered Calmodulin Alleviates Ventricular Arrhythmias in a Calsequestrin-Associated Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. J. Am. Heart Assoc. 2018, 7, e008155. [Google Scholar] [CrossRef]
- Yue, X.; Hazan, A.; Lotteau, S.; Zhang, R.; Torrente, A.G.; Philipson, K.D.; Ottolia, M.; Goldhaber, J.I. Na/Ca exchange in the atrium: Role in sinoatrial node pacemaking and excitation-contraction coupling. Cell Calcium 2020, 87, 102167. [Google Scholar] [CrossRef]
- Li, M.C.H.; O’Brien, T.J.; Todaro, M.; Powell, K.L. Acquired cardiac channelopathies in epilepsy: Evidence, mechanisms, and clinical significance. Epilepsia 2019, 60, 1753–1767. [Google Scholar] [CrossRef]
- Zang, Y.L.; Xia, L. Cellular mechanism of cardiac alternans: An unresolved chicken or egg problem. J. Zhejiang Univ. Sci. B 2014, 15, 201–211. [Google Scholar] [CrossRef]
- Ezeani, M.; Prabhu, S. PI3K(p110α) as a determinant and gene therapy for atrial enlargement in atrial fibrillation. Mol. Cell. Biochem. 2022, 478, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Potenza, D.M.; Janicek, R.; Fernandez-Tenorio, M.; Camors, E.; Ramos-Mondragón, R.; Valdivia, H.H.; Niggli, E. Phosphorylation of the ryanodine receptor 2 at serine 2030 is required for a complete β-adrenergic response. J. Gen. Physiol. 2019, 151, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Hulsurkar, M.M.; Lahiri, S.K.; Karch, J.; Wang, M.C.; Wehrens, X.H.T. Targeting calcium-mediated inter-organellar crosstalk in cardiac diseases. Expert Opin. Ther. Targets 2022, 26, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, X.; Gong, Y.; Zhang, J.; Zang, Y.; Xia, L. Exploring Impaired SERCA Pump-Caused Alternation Occurrence in Ischemia. Comput. Math. Methods Med. 2019, 2019, 8237071. [Google Scholar] [CrossRef]
- Papa, A.; Kushner, J.; Marx, S.O. Adrenergic Regulation of Calcium Channels in the Heart. Annu. Rev. Physiol. 2022, 84, 285–306. [Google Scholar] [CrossRef]
- Chopra, N.; Knollmann, B.C. Triadin regulates cardiac muscle couplon structure and microdomain Ca2+ signalling: A path towards ventricular arrhythmias. Cardiovasc. Res. 2013, 98, 187–191. [Google Scholar] [CrossRef]
- Li, L.; Mirza, S.; Richardson, S.J.; Gallant, E.M.; Thekkedam, C.; Pace, S.M.; Zorzato, F.; Liu, D.; Beard, N.A.; Dulhunty, A.F. A new cytoplasmic interaction between junctin and ryanodine receptor Ca2+ release channels. J. Cell Sci. 2015, 128, 951–963. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, L.; Han, L.; Wang, Y.; Zhou, Y.; Li, Q.; Wu, Y.; Talabieke, S.; Hou, Y.; Wu, L.; et al. Functional Calsequestrin-1 Is Expressed in the Heart and Its Deficiency Is Causally Related to Malignant Hyperthermia-Like Arrhythmia. Circulation 2021, 144, 788–804. [Google Scholar] [CrossRef]
- Brandenburg, S.; Pawlowitz, J.; Steckmeister, V.; Subramanian, H.; Uhlenkamp, D.; Scardigli, M.; Mushtaq, M.; Amlaz, S.I.; Kohl, T.; Wegener, J.W.; et al. A junctional cAMP compartment regulates rapid Ca2+ signaling in atrial myocytes. J. Mol. Cell. Cardiol. 2022, 165, 141–157. [Google Scholar] [CrossRef]
- Xiong, J.; Liu, X.; Gong, Y.; Zhang, P.; Qiang, S.; Zhao, Q.; Guo, R.; Qian, Y.; Wang, L.; Zhu, L.; et al. Pathogenic mechanism of a catecholaminergic polymorphic ventricular tachycardia causing-mutation in cardiac calcium release channel RyR2. J. Mol. Cell. Cardiol. 2018, 117, 26–35. [Google Scholar] [CrossRef]
- Heijman, J.; Ghezelbash, S.; Wehrens, X.H.; Dobrev, D. Serine/Threonine Phosphatases in Atrial Fibrillation. J. Mol. Cell. Cardiol. 2017, 103, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Klapproth, E.; Kämmerer, S.; El-Armouche, A. Function and regulation of phosphatase 1 in healthy and diseased heart. Cell Signal. Cell. Signal. 2022, 90, 110203. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A.; Iravanian, S.; Dolmatova, E.; Jiao, Z.; Liu, H.; Zandieh, S.; Kumar, V.; Wang, K.; Bernstein, K.E.; Bonini, M.G.; et al. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J. Am. Coll. Cardiol. 2011, 58, 2332–2339. [Google Scholar] [CrossRef] [PubMed]
- Potenza, D.M.; Janicek, R.; Fernandez-Tenorio, M.; Niggli, E. Activation of endogenous protein phosphatase 1 enhances the calcium sensitivity of the ryanodine receptor type 2 in murine ventricular cardiomyocytes. J. Physiol. 2020, 598, 1131–1150. [Google Scholar] [CrossRef] [PubMed]
- Said, M.; Becerra, R.; Valverde, C.A.; Kaetzel, M.A.; Dedman, J.R.; Mundiña-Weilenmann, C.; Wehrens, X.H.; Vittone, L.; Mattiazzi, A. Calcium-calmodulin dependent protein kinase II (CaMKII): A main signal responsible for early reperfusion arrhythmias. J. Mol. Cell. Cardiol. 2011, 51, 936–944. [Google Scholar] [CrossRef]
- Bers, D.M.; Grandi, E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J. Cardiovasc. Pharmacol. 2009, 54, 180–187. [Google Scholar] [CrossRef]
- Chang, S.L.; Chuang, H.L.; Chen, Y.C.; Kao, Y.H.; Lin, Y.K.; Yeh, Y.H.; Chen, S.A.; Chen, Y.J. Heart failure modulates electropharmacological characteristics of sinoatrial nodes. Exp. Ther. Med. 2017, 13, 771–779. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Liu, Y.; Zhang, J.; Zheng, X.; Sun, X.; Lei, S.; Kang, Z.; Chen, X.; Lei, M.; et al. Distinct roles of calmodulin and Ca2+/calmodulin-dependent protein kinase II in isopreterenol-induced cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2020, 526, 960–966. [Google Scholar] [CrossRef]
- Yang, H.Q.; Wang, L.P.; Gong, Y.Y.; Fan, X.X.; Zhu, S.Y.; Wang, X.T.; Wang, Y.P.; Li, L.L.; Xing, X.; Liu, X.X.; et al. β2-Adrenergic Stimulation Compartmentalizes β1- Signaling Into Nanoscale Local Domains by Targeting the C-Terminus of β1-Adrenoceptors. Circ. Res. 2019, 124, 1350–1359. [Google Scholar] [CrossRef]
- Demydenko, K.; Sipido, K.R.; Roderick, H.L. Ca2+ release via InsP3Rs enhances RyR recruitment during Ca2+ transients by increasing dyadic [Ca2+] in cardiomyocytes. J. Cell Sci. 2021, 134, jcs258671. [Google Scholar] [CrossRef]
- Jin, X.; Amoni, M.; Gilbert, G.; Dries, E.; Doñate Puertas, R.; Tomar, A.; Nagaraju, C.K.; Pradhan, A.; Yule, D.I.; Martens, T.; et al. InsP3R-RyR Ca2+ channel crosstalk facilitates arrhythmias in the failing human ventricle. Basic Res. Cardiol. 2022, 117, 60. [Google Scholar] [CrossRef]
- Wehrens, X.H.; Lehnart, S.E.; Huang, F.; Vest, J.A.; Reiken, S.R.; Mohler, P.J.; Sun, J.; Guatimosim, S.; Song, L.S.; Rosemblit, N.; et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003, 113, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Taur, Y.; Frishman, W.H. The cardiac ryanodine receptor (RyR2) and its role in heart disease. Cardiol. Rev. 2005, 13, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Dridi, H.; Kushnir, A.; Zalk, R.; Yuan, Q.; Melville, Z.; Marks, A.R. Intracellular calcium leak in heart failure and atrial fibrillation: A unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 2020, 17, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Abramova, T.O. Gene RYR2: GENOKARTA Genetic Encyclopedia. 2020. Available online: https://www.genokarta.ru/gene/RYR2 (accessed on 24 February 2023).
- Napolitano, C.; Mazzanti, A.; Priori, S.G. Genetic risk stratification in cardiac arrhythmias. Curr. Opin. Cardiol. 2018, 33, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Wleklinski, M.J.; Kannankeril, P.J.; Knollmann, B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 2020, 598, 2817–2834. [Google Scholar] [CrossRef] [PubMed]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef]
- Himmel, N.J.; Cox, D.N. Transient receptor potential channels: Current perspectives on evolution, structure, function and nomenclature. Proc. Biol. Sci. 2020, 287, 20201309. [Google Scholar] [CrossRef]
- Samanta, A.; Hughes, T.E.T.; Moiseenkova-Bell, V.Y. Transient Receptor Potential (TRP) Channels. Subcell Biochem. 2018, 87, 141–165. [Google Scholar] [CrossRef]
- Freichel, M.; Berlin, M.; Schürger, A.; Mathar, I.; Bacmeister, L.; Medert, R.; Frede, W.; Marx, A.; Segin, S.; Londoño, J.E.C. TRP Channels in the Heart. In Neurobiology of TRP Channels; Emir, T.L.R., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2017. [Google Scholar]
- Eder, P.; Molkentin, J.D. TRPC channels as effectors of cardiac hypertrophy. Circ. Res. 2011, 108, 265–272. [Google Scholar] [CrossRef]
- Dragún, M.; Gažová, A.; Kyselovič, J.; Hulman, M.; Máťuš, M. TRP Channels Expression Profile in Human End-Stage Heart Failure. Medicina 2019, 55, 380. [Google Scholar] [CrossRef] [PubMed]
- Falcón, D.; Galeano-Otero, I.; Martín-Bórnez, M.; Fernández-Velasco, M.; Gallardo- Castillo, I.; Rosado, J.A.; Ordóñez, A.; Smani, T. TRPC Channels: Dysregulation and Ca2+/Mishandling in Ischemic Heart Disease. Cells 2020, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Inoue, R.; Kurahara, L.H.; Hiraishi, K. TRP channels in cardiac and intestinal fibrosis. Semin. Cell Dev. Biol. 2019, 94, 40–49. [Google Scholar] [CrossRef]
- Yue, Z.; Zhang, Y.; Xie, J.; Jiang, J.; Yue, L. Transient receptor potential (TRP) channels and cardiac fibrosis. Curr. Top. Med. Chem. 2013, 13, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Numaga-Tomita, T.; Nishida, M. TRPC Channels in Cardiac Plasticity. Cells 2020, 9, 454. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Wilkin, B.J.; Bodi, I.; Molkentin, J.D. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006, 20, 1660–1670. [Google Scholar] [CrossRef]
- Satoh, S.; Tanaka, H.; Ueda, Y.; Oyama, J.; Sugano, M.; Sumimoto, H.; Mori, Y.; Makino, N. Transient receptor potential (TRP) protein 7 acts as a G protein-activated Ca2+ channel mediating angiotensin II-induced myocardial apoptosis. Mol. Cell. Biochem. 2007, 294, 205–215. [Google Scholar] [CrossRef]
- Liao, Y.; Plummer, N.W.; George, M.D.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC: Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. USA 2009, 106, 3202–3206. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef]
- Dyrda, A.; Koenig, S.; Frieden, M. STIM1 long and STIM1 gate differently TRPC1 during store-operated calcium entry. Cell Calcium 2020, 86, 102134. [Google Scholar] [CrossRef]
- Kühn, F.J.; Heiner, I.; Lückhoff, A. TRPM2: A calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflug. Arch. 2005, 451, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Ru, X.; Yao, X. TRPM2: A multifunctional ion channel for oxidative stress sensing. Acta Physiol. Sin. 2014, 66, 7–15. [Google Scholar] [PubMed]
- Cheung, J.Y.; Miller, B.A. Transient Receptor Potential-Melastatin Channel Family Member 2: Friend or Foe. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 308–329. [Google Scholar]
- Kruse, M.; Schulze-Bahr, E.; Corfield, V.; Beckmann, A.; Stallmeyer, B.; Kurtbay, G.; Ohmert, I.; Schulze-Bahr, E.; Brink, P.; Pongs, O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Investig. 2009, 119, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zong, P.; Yan, J.; Yue, Z.; Li, X.; Smith, C.; Ai, X.; Yue, L. Upregulation of transient receptor potential melastatin 4 (TRPM4) in ventricular fibroblasts from heart failure patients. Pflug. Arch. 2021, 473, 521–531. [Google Scholar] [CrossRef]
- Dong, Y.; Du, R.; Fan, L.L.; Jin, J.Y.; Huang, H.; Chen, Y.Q.; Bi, D.D.; Xiang, R. Whole-Exome Sequencing Identifies a Novel TRPM4 Mutation in a Chinese Family with Atrioventricular Block. Biomed. Res. Int. 2021, 17, 9247541. [Google Scholar] [CrossRef]
- Hu, Y.; Li, Q.; Kurahara, L.H.; Shioi, N.; Hiraishi, K.; Fujita, T.; Zhu, X.; Inoue, R. An Arrhythmic Mutation E7K Facilitates TRPM4 Channel Activation via Enhanced PIP2 Interaction. Cells 2021, 10, 983. [Google Scholar] [CrossRef]
- Sah, R.; Mesirca, P.; Mason, X.; Gibson, W.; Bates-Withers, C.; Van den Boogert, M.; Chaudhuri, D.; Pu, W.T.; Mangoni, M.E.; Clapham, D.E. Timing of myocardial trpm7 deletion during cardiogenesis variably disrupts adult ventricular function, conduction, and repolarization. Circulation 2013, 128, 101–114. [Google Scholar] [CrossRef]
- Cartwright, J.H.; Aziz, Q.; Harmer, S.C.; Thayyil, S.; Tinker, A.; Munroe, P.B. Genetic variants in TRPM7 associated with unexplained stillbirth modify ion channel function. Hum. Mol. Genet. 2020, 29, 1797–1807. [Google Scholar] [CrossRef]
- Conklin, D.J.; Guo, Y.; Nystoriak, M.A.; Jagatheesan, G.; Obal, D.; Kilfoil, P.J.; Hoetker, J.D.; Guo, L.; Bolli, R.; Bhatnagar, A. TRPA1 channel contributes to myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H889–H899. [Google Scholar] [CrossRef]
- Martín-Bórnez, M.; Galeano-Otero, I.; Del Toro, R.; Smani, T. TRPC and TRPV Channels’ Role in Vascular Remodeling and Disease. Int. J. Mol. Sci. 2020, 21, 6125. [Google Scholar] [CrossRef] [PubMed]
- Üstünel, L.; Özgüler, I.M. The effects of iloprost and beta3 receptor agonist on TRPA1 and TRPC1 immunreactivity in an experimental lower extremty ischemia-reperfusion injury model. Turk. J. Med. Sci. 2021, 51, 2763–2770. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Kanematsu, M.; Sakai, K.; Matsuda, T.; Hattori, N.; Mizuno, Y.; Suzuki, D.; Miyata, T.; Noguchi, N.; Niki, E.; et al. Protein-bound acrolein: Potential markers for oxidative stress. Proc. Natl. Acad. Sci. USA 1998, 95, 4882–4887. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, L.J.; Dubin, A.E.; Evans, M.J.; Marr, F.; Schultz, P.G.; Cravatt, B.F.; Patapoutian, A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545. [Google Scholar] [CrossRef]
- Hu, Q.; Ahmad, A.A.; Seidel, T.; Hunter, C.; Streiff, M.; Nikolova, L.; Spitzer, K.W.; Sachse, F.B. Location and function of transient receptor potential canonical channel 1 in ventricular myocytes. J. Mol. Cell. Cardiol. 2020, 139, 113–123. [Google Scholar] [CrossRef]
- Firth, A.L.; Remillard, C.V.; Yuan, J.X. TRP channels in hypertension. Biochim. Biophys. Acta 2007, 1772, 895–906. [Google Scholar] [CrossRef]
- Yue, Z.; Xie, J.; Yu, A.S.; Stock, J.; Du, J.; Yue, L. Role of TRP Channels in the Cardiovascular System. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H157–H182. [Google Scholar] [CrossRef]
- Andriulė, I.; Pangonytė, D.; Gwanyanya, A.; Karčiauskas, D.; Mubagwa, K.; Mačianskienė, R. Detection of TRPM6 and TRPM7 Proteins in Normal and Diseased Cardiac Atrial Tissue and Isolated Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 14860. [Google Scholar] [CrossRef]
- Bogdanova, E.; Beresneva, O.; Galkina, O.; Zubina, I.; Ivanova, G.; Parastaeva, M.; Semenova, N.; Dobronravov, V. Myocardial Hypertrophy and Fibrosis Are Associated with Cardiomyocyte Beta-Catenin and TRPC6/Calcineurin/NFAT Signaling in Spontaneously Hypertensive Rats with 5/6 Nephrectomy. Int. J. Mol. Sci. 2021, 22, 4645. [Google Scholar] [CrossRef]
- Onyali, V.C.; Domeier, T.L. Cardiac TRPV4 channels. Curr. Top. Membr. 2022, 89, 63–74. [Google Scholar] [CrossRef]
- Hong, K.S.; Lee, M.G. Endothelial Ca2+ signaling-dependent vasodilation through transient receptor potential channels. Korean J. Physiol. Pharmacol. 2020, 24, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Kochukov, M.Y.; Balasubramanian, A.; Noel, R.C.; Marrelli, S.P. Role of TRPC1 and TRPC3 channels in contraction and relaxation of mouse thoracic aorta. J. Vasc. Res. 2013, 50, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Willette, R.N.; Bao, W.; Nerurkar, S.; Yue, T.L.; Doe, C.P.; Stankus, G.; Turner, G.H.; Ju, H.; Thomas, H.; Fishman, C.E.; et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J. Pharmacol. Exp. Ther. 2008, 326, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Heppner, T.J.; Nelson, M.T.; Brayden, J.E. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ. Res. 2005, 97, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Miguel, I.; Cidad, P.; Pérez-García, M.T.; López-López, J.R. Differences in TRPC3 and TRPC6 channels assembly in mesenteric vascular smooth muscle cells in essential hypertension. J. Physiol. 2017, 595, 1497–1513. [Google Scholar] [CrossRef]
- Mathar, I.; Vennekens, R.; Meissner, M.; Kees, F.; Van der Mieren, G.; Camacho Londoño, J.E.; Uhl, S.; Voets, T.; Hummel, B.; van den Bergh, A.; et al. Increased catecholamine secretion contributes to hypertension in TRPM4-deficient mice. J. Clin. Investig. 2010, 120, 3267–3279. [Google Scholar] [CrossRef]
- Wei, J.; Ching, L.C.; Zhao, J.F.; Shyue, S.K.; Lee, H.F.; Kou, Y.R.; Lee, T.S. Essential Role of Transient Receptor Potential Vanilloid Type 1 in Evodiamine-Mediated Protection Against Atherosclerosis. Acta Physiol. 2013, 207, 299–307. [Google Scholar] [CrossRef]
- Nishida, M.; Tanaka, T.; Mangmool, S.; Nishiyama, K.; Nishimura, A. Canonical Transient Receptor Potential Channels and Vascular Smooth Muscle Cell Plasticity. J. Lipid Atheroscler. 2020, 9, 124–139. [Google Scholar] [CrossRef]
- Mukherjee, P.; Rahaman, S.G.; Goswami, R.; Dutta, B.; Mahanty, M.; Rahaman, S.O. Role of mechanosensitive channels/receptors in atherosclerosis. Am. J. Physiol.-Cell Physiol. 2022, 322, C927–C938. [Google Scholar] [CrossRef]
- Liu, D.; Zhu, Z.; Tepel, M. The role of transient receptor potential channels in metabolic syndrome. Hypertens. Res. 2008, 31, 1989–1995. [Google Scholar] [CrossRef][Green Version]
- Nishiyama, K.; Tanaka, T.; Nishimura, A.; Nishida, M. TRPC3-Based Protein Signaling Complex as a Therapeutic Target of Myocardial Atrophy. Curr. Mol. Pharmacol. 2021, 14, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Hiraishi, K.; Kurahara, L.H.; Ishikawa, K.; Go, T.; Yokota, N.; Hu, Y.; Fujita, T.; Inoue, R.; Hirano, K. Potential of the TRPM7 channel as a novel therapeutic target for pulmonary arterial hypertension. J. Smooth Muscle Res. 2022, 58, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, A.; McCormick, D.A. H-current: Properties of a neuronal and network pacemaker. Neuron 1998, 21, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Hennis, K.; Biel, M.; Wahl-Schott, C.; Fenske, S. Beyond pacemaking: HCN channels in sinoatrial node function. Prog. Biophys. Mol. Biol. 2021, 166, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, A.; Bauer, D.; Giese, M.H.; Swuec, P.; Porro, A.; Gasparri, F.; Sharifzadeh, A.S.; Chaves-Sanjuan, A.; Alberio, L.; Parisi, G.; et al. Gating movements and ion permeation in HCN4 pacemaker channels. Mol. Cell 2021, 81, 2929–2943. [Google Scholar] [CrossRef] [PubMed]
- Santoro, B.; Tibbs, G.R. The HCN gene family: Molecular basis of the hyperpolarization-activated pacemaker channels. Ann. N. Y. Acad. Sci. 1999, 868, 741–764. [Google Scholar] [CrossRef]
- Biel, M.; Schneider, A.; Wahl, C. Cardiac HCN channels: Structure, function, and modulation. Trends Cardiovasc. Med. 2002, 12, 206–212. [Google Scholar] [CrossRef]
- Cohen, I.S.; Robinson, R.B. Pacemaker current and automatic rhythms: Toward a molecular understanding. Handb. Exp. Pharmacol. 2006, 171, 41–71. [Google Scholar] [CrossRef]
- Farraha, M.; Kumar, S.; Chong, J.; Cho, H.C.; Kizana, E. Gene Therapy Approaches to Biological Pacemakers. J. Cardiovasc. Dev. Dis. 2018, 5, 50. [Google Scholar] [CrossRef]
- Van Wagoner, D.R.; Lindsay, B.D. Phosphodiesterase-4 activity: A critical modulator of atrial contractility and arrhythmogenesis. J. Am. Coll. Cardiol. 2012, 59, 2191–2192. [Google Scholar] [CrossRef]
- Yu, X.; Chen, X.W.; Zhou, P.; Yao, L.; Liu, T.; Zhang, B.; Li, Y.; Zheng, H.; Zheng, L.H.; Zhang, C.X.; et al. Calcium influx through If channels in rat ventricular myocytes. Am. J. Physiol.-Cell Physiol. 2007, 292, C1147–C1155. [Google Scholar] [CrossRef] [PubMed]
- Michels, G.; Brandt, M.C.; Zagidullin, N.; Khan, I.F.; Larbig, R.; van Aaken, S.; Wippermann, J.; Hoppe, U.C. Direct evidence for calcium conductance of hyperpolarization-activated cyclic nucleotide-gated channels and human native If at physiological calcium concentrations. Cardiovasc. Res. 2008, 78, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Ohtani, K.; Higo, T.; Tanaka, M.; Kawasaki, Y.; Tsutsui, H. Ivabradine for the treatment of cardiovascular diseases. Circ. J. 2019, 83, 252–260. [Google Scholar] [CrossRef]
- Wang, Z.J.; Blanco, I.; Hayoz, S.; Brelidze, T.I. The HCN domain is required for HCN channel cell-surface expression and couples voltage- and cAMP-dependent gating mechanisms. J. Biol. Chem. 2020, 295, 8164–8173. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Laird, J.G.; Yamaguchi, D.M.; Baker, S.A. An N-terminal ER export signal facilitates the plasma membrane targeting of HCN1 channels in photoreceptors. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3514–3521. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Proenza, C.; Tran, N.; Angoli, D.; Zahynacz, K.; Balcar, P.; Accili, E.A. Different roles for the cyclic nucleotide binding domain and amino terminus in assembly and expression of hyperpolarization-activated, cyclic nucleotide-gated channels. J. Biol. Chem. 2002, 277, 29634–29642. [Google Scholar] [CrossRef]
- Shimizu, M.; Mi, X.; Toyoda, F.; Kojima, A.; Ding, W.G.; Fukushima, Y.; Omatsu-Kanbe, M.; Kitagawa, H.; Matsuura, H. Propofol, an Anesthetic Agent, Inhibits HCN Channels through the Allosteric Modulation of the cAMP-Dependent Gating Mechanism. Biomolecules 2022, 12, 570. [Google Scholar] [CrossRef]
- Baruscotti, M.; Bucchi, A.; Difrancesco, D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol. Ther. 2005, 107, 59–79. [Google Scholar] [CrossRef]
- Hennis, K.; Rötzer, R.D.; Piantoni, C.; Biel, M.; Wahl-Schott, C.; Fenske, S. Speeding Up the Heart? Traditional and New Perspectives on HCN4 Function. Front. Physiol. 2021, 12, 669029. [Google Scholar] [CrossRef]
- Wahl-Schott, C.; Biel, M. HCN channels: Structure, cellular regulation and physiological function. Cell. Mol. Life Sci. 2009, 66, 470–494. [Google Scholar] [CrossRef]
- Scicchitano, P.; Carbonara, S.; Ricci, G.; Mandurino, C.; Locorotondo, M.; Bulzis, G.; Gesualdo, M.; Zito, A.; Carbonara, R.; Dentamaro, I.; et al. HCN channels and heart rate. Molecules 2012, 17, 4225–4235. [Google Scholar] [CrossRef] [PubMed]
- Hummert, S.; Thon, S.; Eick, T.; Schmauder, R.; Schulz, E.; Benndorf, K. Activation gating in HCN2 channels. PLoS Comput. Biol. 2018, 14, e1006045. [Google Scholar] [CrossRef] [PubMed]
- VanSchouwen, B.; Melacini, G. Regulation of HCN Ion Channels by Non-canonical Cyclic Nucleotides. Handb. Exp. Pharmacol. 2017, 238, 123–133. [Google Scholar] [CrossRef]
- Peters, C.H.; Singh, R.K.; Bankston, J.R.; Proenza, C. Regulation of HCN Channels by Protein Interactions. Front. Physiol. 2022, 13, 928507. [Google Scholar] [CrossRef] [PubMed]
- Erlenhardt, N.; Kletke, O.; Wohlfarth, F.; Komadowski, M.A.; Clasen, L.; Makimoto, H.; Rinné, S.; Kelm, M.; Jungen, C.; Decher, N.; et al. Disease-associated HCN4 V759I variant is not sufficient to impair cardiac pacemaking. Pflug. Arch. 2020, 472, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Nakamura, K.; Hisatomi, Y.; Matsumoto, S.; Suzuki, M.; Harvey, R.P.; Kurihara, H.; Hattori, S.; Yamamoto, T.; Michalak, M.; et al. Arrhythmia induced by spatiotemporal overexpression of calreticulin in the heart. Mol. Genet. Metab. 2007, 91, 285–293. [Google Scholar] [CrossRef]
- Saeed, Y.; Temple, I.P.; Borbas, Z.; Atkinson, A.; Yanni, J.; Maczewski, M.; Mackiewicz, U.; Aly, M.; Logantha, S.J.R.J.; Garratt, C.J.; et al. Structural and functional remodeling of the atrioventricular node with aging in rats: The role of hyperpolarization-activated cyclic nucleotide-gated and ryanodine 2 channels. Heart Rhythm 2018, 15, 752–760. [Google Scholar] [CrossRef]
- Rivolta, I.; Binda, A.; Masi, A.; DiFrancesco, J.C. Cardiac and neuronal HCN channelopathies. Pflug. Arch. 2020, 472, 931–951. [Google Scholar] [CrossRef]
- Qu, J.; Plotnikov, A.N.; Danilo, P.; Shlapakova, I.; Cohen, I.S.; Robinson, R.B.; Rosen, M.R. Expression and function of a biological pacemaker in canine heart. Circulation 2003, 107, 1106–1109. [Google Scholar] [CrossRef]
- Lo, Y.; Lin, L.Y.; Tsai, T.F. Use of calcium channel blockers in dermatology: A narrative review. Expert Rev. Clin. Pharmacol. 2021, 14, 481–489. [Google Scholar] [CrossRef]
- Kokilambigai, K.S.; Kavitha, J.; Seetharaman, R.; Lakshmi, K.S.; Sai Susmitha, A. Analytical and Bioanalytical Techniques for the Quantification of the Calcium Channel Blocker—Amlodipine: A Critical Review. Crit. Rev. Anal. Chem. 2021, 51, 754–786. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, D.A.; Ozer, L.Y.; Al Haj Zen, A. A Novel High Content Angiogenesis Assay Reveals That Lacidipine, L-Type Calcium Channel Blocker, Induces In Vitro Vascular Lumen Expansion. Int. J. Mol. Sci. 2022, 23, 4891. [Google Scholar] [CrossRef] [PubMed]
- Rabah, F.; El-Naggari, M.; Al-Nabhani, D. Amlodipine: The double edged sword. J. Paediatr. Child Health 2017, 53, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Kung, J.Y.; Mitchelmore, B.; Cave, A.; Banh, H.L. Comparative peripheral edema for dihydropyridines calcium channel blockers treatment: A systematic review and network meta-analysis. J. Clin. Hypertens. 2022, 24, 536–554. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, R.; Wang, K.; Zhang, W.; Zhang, M.; Jin, L.; Xie, P.; Zheng, W.; Shang, H.; Hu, Q.; et al. Novel CaMKII-δ Inhibitor Hesperadin Exerts Dual Functions to Ameliorate Cardiac Ischemia/Reperfusion Injury and Inhibit Tumor Growth. Circulation 2022, 145, 1154–1168. [Google Scholar] [CrossRef]
- Yao, Y.; Li, F.; Zhang, M.; Jin, L.; Xie, P.; Liu, D.; Zhang, J.; Hu, X.; Lv, F.; Shang, H.; et al. Targeting CaMKII-δ9 Ameliorates Cardiac Ischemia/Reperfusion Injury by Inhibiting Myocardial Inflammation. Circ. Res. 2022, 130, 887–903. [Google Scholar] [CrossRef]
- Xia, H.; Zahra, A.; Jia, M.; Wang, Q.; Wang, Y.; Campbell, S.L.; Wu, J. Na+/H+ Exchanger 1, a Potential Therapeutic Drug Target for Cardiac Hypertrophy and Heart Failure. Pharmaceuticals 2022, 15, 875. [Google Scholar] [CrossRef]
- Kamel, R.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiac hypertrophy and heart failure. Nat. Rev. Cardiol. 2023, 20, 90–108. [Google Scholar] [CrossRef]
- Mengesha, H.G.; Tafesse, T.B.; Bule, M.H. If Channel as an Emerging Therapeutic Target for Cardiovascular Diseases: A Review of Current Evidence and Controversies. Front. Pharmacol. 2017, 8, 874. [Google Scholar] [CrossRef]
- Ma, S.; Jiang, Y.; Huang, W.; Li, X.; Li, S. Role of Transient Receptor Potential Channels in Heart Transplantation: A Potential Novel Therapeutic Target for Cardiac Allograft Vasculopathy. Med. Sci. Monit. 2017, 23, 2340–2347. [Google Scholar] [CrossRef]
- Cheraghi, M.; Negahdari, B.; Daraee, H.; Eatemadi, A. Heart targeted nanoliposomal/nanoparticles drug delivery: An updated review. Biomed. Pharmacother. 2017, 86, 316–323. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).