
Constitutive Interleukin-7 Cytokine Signaling Enhances the Persistence of Epstein–Barr Virus-Specific T-Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
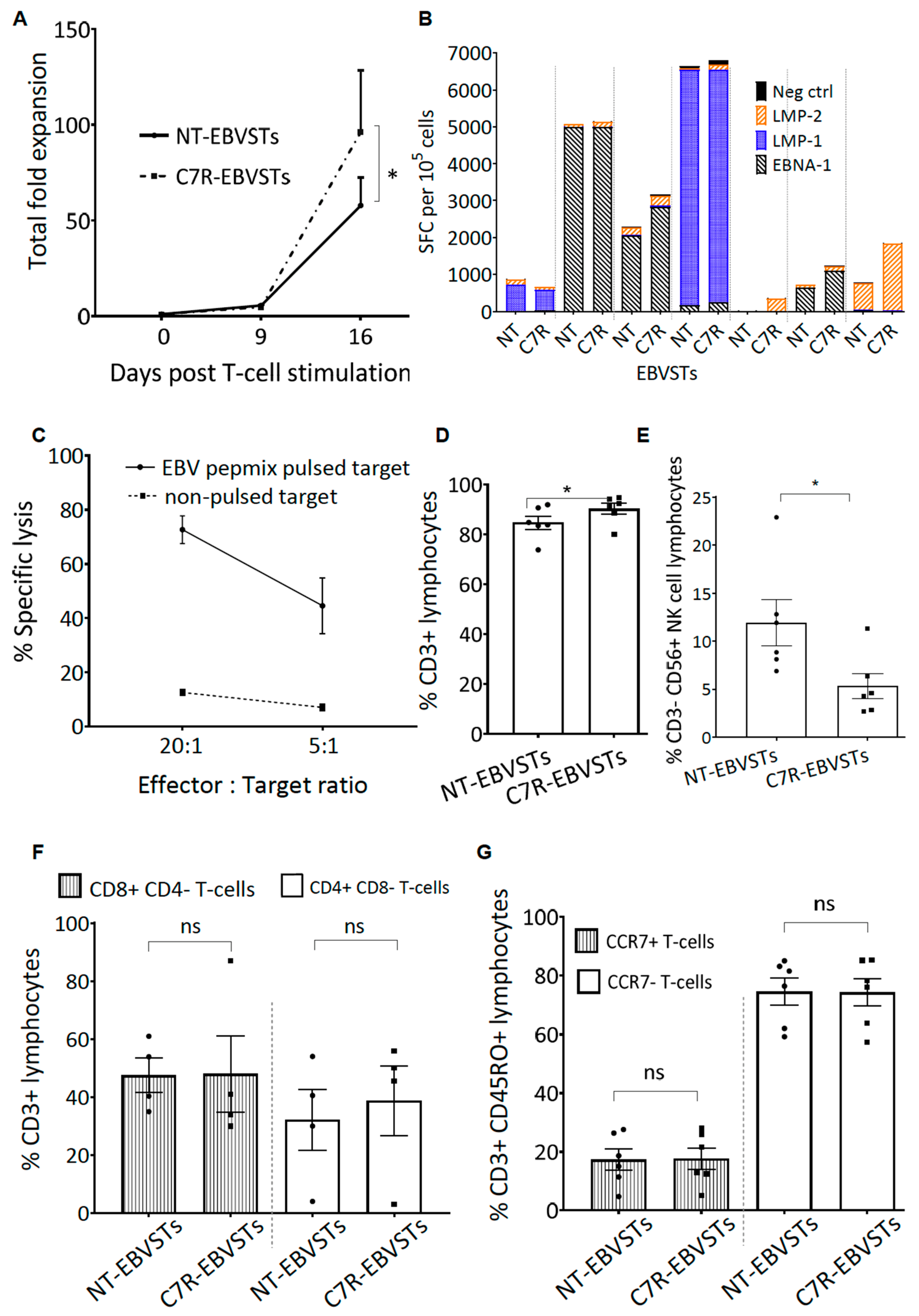
2.1. Constitutive STAT5 Activation in EBVSTs Expressing the Constitutively Active IL7 Receptor (C7R)
2.2. C7R-EBVSTs Maintain EBV Antigen Specificity and Cytotoxic Function
2.3. C7R Enhances the Survival and Specificity of EBVSTs in the Absence of Cytokines
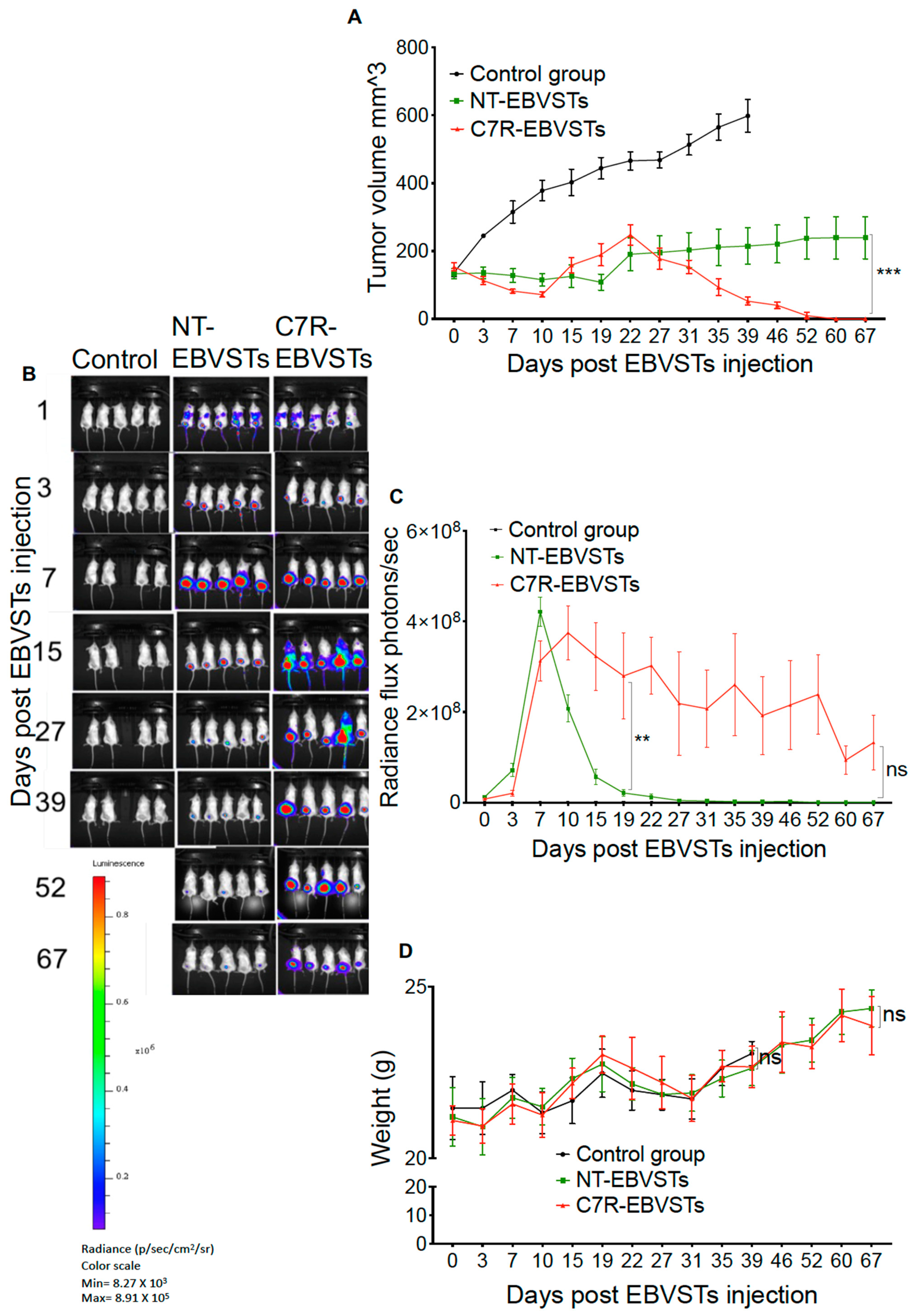
2.4. C7R Increases the Persistence and Anti-Tumor Activity of EBVSTs In-Vivo
3. Discussion
4. Materials and Methods
4.1. Blood Donors and Cell Lines
4.2. CD3 and CD28-Activated T-Cells (ATCs) for Use as Antigen-Presenting Cells (APCs)
4.3. Pepmixes
4.4. Cell Culture Media
4.5. LCL Generation
4.6. Costimulatory Cell Lines
4.7. Generation of Retroviral Vectors
4.8. Generation of Irradiated, Antigen-Presenting Cell Complex for the Second and Subsequent Stimulations of EBVSTs
4.9. EBVST Generation
4.10. Transduction of EBVSTs with C7R
4.11. Immunophenotyping
4.12. Phosphorylated-STAT5 Assay
4.13. Enzyme-Linked Immunospot (ELISpot) Assay
4.14. Cytotoxicity Assay
4.15. In-Vivo Murine Model
4.16. Statistical Analysis
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, L.S.; Rickinson, A.B. Epstein–Barr Virus: 40 Years On. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr Virus: More than 50 Years Old and Still Providing Surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Münz, C. Latency and Lytic Replication in Epstein–Barr Virus-Associated Oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E. Use of Gene-Modified Virus-Specific T Lymphocytes to Control Epstein–Barr-Virus-Related Lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.-F.; Rochester, R.J.; et al. Long-Term Outcome of EBV-Specific T-Cell Infusions to Prevent or Treat EBV-Related Lymphoproliferative Disease in Transplant Recipients. Blood 2010, 115, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Woods, M.; Mehta, N.U.; Sauer, T.; Parikh, K.S.; Schmuck-Henneresse, M.; Zhang, H.; Mehta, B.; Brenner, M.K.; Heslop, H.E.; et al. Naive T Cells Inhibit the Outgrowth of Intractable Antigen-Activated Memory T Cells: Implications for T-Cell Immunotherapy. J. Immunother. Cancer 2023, 11, e006267. [Google Scholar] [CrossRef]
- Bollard, C.M.; Straathof, K.C.M.; Huls, M.H.; Leen, A.; Lacuesta, K.; Davis, A.; Gottschalk, S.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. The Generation and Characterization of LMP2-Specific CTLs for Use as Adoptive Transfer From Patients With Relapsed EBV-Positive Hodgkin Disease. J. Immunother. 2004, 27, 317–327. [Google Scholar] [CrossRef]
- Bollard, C.M.; Huls, M.H.; Buza, E.; Weiss, H.; Torrano, V.; Gresik, M.V.; Chang, J.; Gee, A.; Gottschalk, S.M.; Carrum, G.; et al. Administration of Latent Membrane Protein 2–Specific Cytotoxic T Lymphocytes to Patients with Relapsed Epstein–Barr Virus–Positive Lymphoma. Clin. Lymphoma Myeloma 2006, 6, 342–347. [Google Scholar] [CrossRef]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained Complete Responses in Patients with Lymphoma Receiving Autologous Cytotoxic T Lymphocytes Targeting Epstein–Barr Virus Latent Membrane Proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The Immunosuppressive Tumour Network: Myeloid-Derived Suppressor Cells, Regulatory T Cells and Natural Killer T Cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T Cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef]
- de la Cruz-Merino, L.; Lejeune, M.; Nogales Fernández, E.; Henao Carrasco, F.; Grueso López, A.; Illescas Vacas, A.; Pulla, M.P.; Callau, C.; Álvaro, T. Role of Immune Escape Mechanisms in Hodgkin’s Lymphoma Development and Progression: A Whole New World with Therapeutic Implications. Clin. Dev. Immunol. 2012, 2012, 756353. [Google Scholar] [CrossRef]
- Mocellin, S.; Wang, E.; Marincola, F.M. Cytokines and Immune Response in the Tumor Microenvironment. J. Immunother. 2001, 24, 392–407. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Grote, D.M.; Ziesmer, S.C.; Xiu, B.; Yates, N.R.; Secreto, F.J.; Hodge, L.S.; Witzig, T.E.; Novak, A.J.; Ansell, S.M. Soluble and Membrane-Bound TGF-β-Mediated Regulation of Intratumoral T Cell Differentiation and Function in B-Cell Non-Hodgkin Lymphoma. PLoS ONE 2013, 8, e59456. [Google Scholar] [CrossRef]
- Aldinucci, D.; Borghese, C.; Casagrande, N. Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It. Int. J. Mol. Sci. 2019, 20, 2416. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, S.; Sun, R.; Wu, T.; Qian, J. Understanding the Interplay between Host Immunity and Epstein–Barr Virus in NPC Patients. Emerg. Microbes Infect. 2015, 4, e20. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Dudley, M.E. Adoptive Cell Therapy for the Treatment of Patients with Metastatic Melanoma. Curr. Opin. Immunol. 2009, 21, 233–240. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, Hyperproliferation, Activation of Natural Killer Cells and CD8 T Cells, and Cytokine Production During First-in-Human Clinical Trial of Recombinant Human Interleukin-15 in Patients With Cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of Single-Dose Interleukin-12 Exposure on Interleukin-12-Associated Toxicity and Interferon-Gamma Production. Blood 1997, 90, 2541–2548. [Google Scholar]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive Cell Therapy for Patients With Metastatic Melanoma: Evaluation of Intensive Myeloablative Chemoradiation Preparative Regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.; Khong, H.; Antony, P.; Palmer, D.; Restifo, N. Sinks, Suppressors and Antigen Presenters: How Lymphodepletion Enhances T Cell-Mediated Tumor Immunotherapy. Trends Immunol. 2005, 26, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. JNCI J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-Term Outcomes Following CAR T Cell Therapy: What We Know so Far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-Infiltrating Lymphocytes Genetically Engineered with an Inducible Gene Encoding Interleukin-12 for the Immunotherapy of Metastatic Melanoma. Clin. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Xu, X.; Courtney, A.N.; Tian, G.; Barragan, G.A.; Guo, L.; Amador, C.M.; Ghatwai, N.; Rathi, P.; Wood, M.S.; et al. Anti-GD2 CAR-NKT Cells in Relapsed or Refractory Neuroblastoma: Updated Phase 1 Trial Interim Results. Nat. Med. 2023, 29, 1379–1388. [Google Scholar] [CrossRef]
- Lange, S.; Sand, L.G.L.; Bell, M.; Patil, S.L.; Langfitt, D.; Gottschalk, S. A Chimeric GM-CSF/IL18 Receptor to Sustain CAR T-Cell Function. Cancer Discov. 2021, 11, 1661–1671. [Google Scholar] [CrossRef]
- Tang, L.; Zheng, Y.; Melo, M.B.; Mabardi, L.; Castaño, A.P.; Xie, Y.-Q.; Li, N.; Kudchodkar, S.B.; Wong, H.C.; Jeng, E.K.; et al. Enhancing T Cell Therapy through TCR-Signaling-Responsive Nanoparticle Drug Delivery. Nat. Biotechnol. 2018, 36, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-Function Mutations in Interleukin-7 Receptor-α (IL7R) in Childhood Acute Lymphoblastic Leukemias. J. Exp. Med. 2011, 208, 901–908. [Google Scholar] [CrossRef]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R Gain-of-Function Mutations in Childhood T-Cell Acute Lymphoblastic Leukemia. Nat. Genet. 2011, 43, 932–939. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. Biomed. Res. Int. 2020, 2020, 4795171. [Google Scholar] [CrossRef] [PubMed]
- Czerkinsky, C.; Andersson, G.; Ekre, H.-P.; Nilsson, L.-Å.; Klareskog, L.; Ouchterlony, Ö. Reverse ELISPOT Assay for Clonal Analysis of Cytokine Production I. Enumeration of Gamma-Interferon-Secreting Cells. J. Immunol. Methods 1988, 110, 29–36. [Google Scholar] [CrossRef]
- Bollard, C.M.; Gottschalk, S.; Leen, A.M.; Weiss, H.; Straathof, K.C.; Carrum, G.; Khalil, M.; Wu, M.; Huls, M.H.; Chang, C.-C.; et al. Complete Responses of Relapsed Lymphoma Following Genetic Modification of Tumor-Antigen Presenting Cells and T-Lymphocyte Transfer. Blood 2007, 110, 2838–2845. [Google Scholar] [CrossRef]
- Sportès, C.; Babb, R.R.; Krumlauf, M.C.; Hakim, F.T.; Steinberg, S.M.; Chow, C.K.; Brown, M.R.; Fleisher, T.A.; Noel, P.; Maric, I.; et al. Phase I Study of Recombinant Human Interleukin-7 Administration in Subjects with Refractory Malignancy. Clin. Cancer Res. 2010, 16, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Ebong, S.; Yu, C.-R.; Carper, D.A.; Chepelinsky, A.B.; Egwuagu, C.E. Activation of STAT Signaling Pathways and Induction of Suppressors of Cytokine Signaling (SOCS) Proteins in Mammalian Lens by Growth Factors. Investig. Opthalmology Vis. Sci. 2004, 45, 872. [Google Scholar] [CrossRef] [PubMed]
- Gatzka, M.; Piekorz, R.; Moriggl, R.; Rawlings, J.; Ihle, J.N. A Role for STAT5A/B in Protection of Peripheral T-Lymphocytes from Postactivation Apoptosis: Insights from Gene Expression Profiling. Cytokine 2006, 34, 143–154. [Google Scholar] [CrossRef]
- Cholez, E.; Debuysscher, V.; Bourgeais, J.; Boudot, C.; Leprince, J.; Tron, F.; Brassart, B.; Regnier, A.; Bissac, E.; Pecnard, E.; et al. Evidence for a Protective Role of the STAT5 Transcription Factor against Oxidative Stress in Human Leukemic Pre-B Cells. Leukemia 2012, 26, 2390–2397. [Google Scholar] [CrossRef]
- Savoldo, B.; Huls, M.H.; Liu, Z.; Okamura, T.; Volk, H.-D.; Reinke, P.; Sabat, R.; Babel, N.; Jones, J.F.; Webster-Cyriaque, J.; et al. Autologous Epstein–Barr Virus (EBV)–Specific Cytotoxic T Cells for the Treatment of Persistent Active EBV Infection. Blood 2002, 100, 4059–4066. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Rooney, C.M.; Heslop, H.E. T-Cell Therapy in the Treatment of Post-Transplant Lymphoproliferative Disease. Nat. Rev. Clin. Oncol. 2012, 9, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Ngo, M.C.; Ando, J.; Leen, A.M.; Ennamuri, S.; Lapteva, N.; Vera, J.F.; Min-Venditti, A.; Mims, M.P.; Heslop, H.E.; Bollard, C.M.; et al. Complementation of Antigen-Presenting Cells to Generate T Lymphocytes With Broad Target Specificity. J. Immunother. 2014, 37, 193–203. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Sauer, T.; Omer, B.A.; Shum, T.; Rollins, L.A.; Rooney, C.M. Constitutive Interleukin-7 Cytokine Signaling Enhances the Persistence of Epstein–Barr Virus-Specific T-Cells. Int. J. Mol. Sci. 2023, 24, 15806. https://doi.org/10.3390/ijms242115806
Sharma S, Sauer T, Omer BA, Shum T, Rollins LA, Rooney CM. Constitutive Interleukin-7 Cytokine Signaling Enhances the Persistence of Epstein–Barr Virus-Specific T-Cells. International Journal of Molecular Sciences. 2023; 24(21):15806. https://doi.org/10.3390/ijms242115806
Chicago/Turabian StyleSharma, Sandhya, Tim Sauer, Bilal A. Omer, Thomas Shum, Lisa A. Rollins, and Cliona M. Rooney. 2023. "Constitutive Interleukin-7 Cytokine Signaling Enhances the Persistence of Epstein–Barr Virus-Specific T-Cells" International Journal of Molecular Sciences 24, no. 21: 15806. https://doi.org/10.3390/ijms242115806
APA StyleSharma, S., Sauer, T., Omer, B. A., Shum, T., Rollins, L. A., & Rooney, C. M. (2023). Constitutive Interleukin-7 Cytokine Signaling Enhances the Persistence of Epstein–Barr Virus-Specific T-Cells. International Journal of Molecular Sciences, 24(21), 15806. https://doi.org/10.3390/ijms242115806