
The Biological Assessment of Shikonin and β,β-dimethylacrylshikonin Using a Cellular Myxofibrosarcoma Tumor Heterogeneity Model
 , and
, and
Abstract
:1. Introduction
2. Results
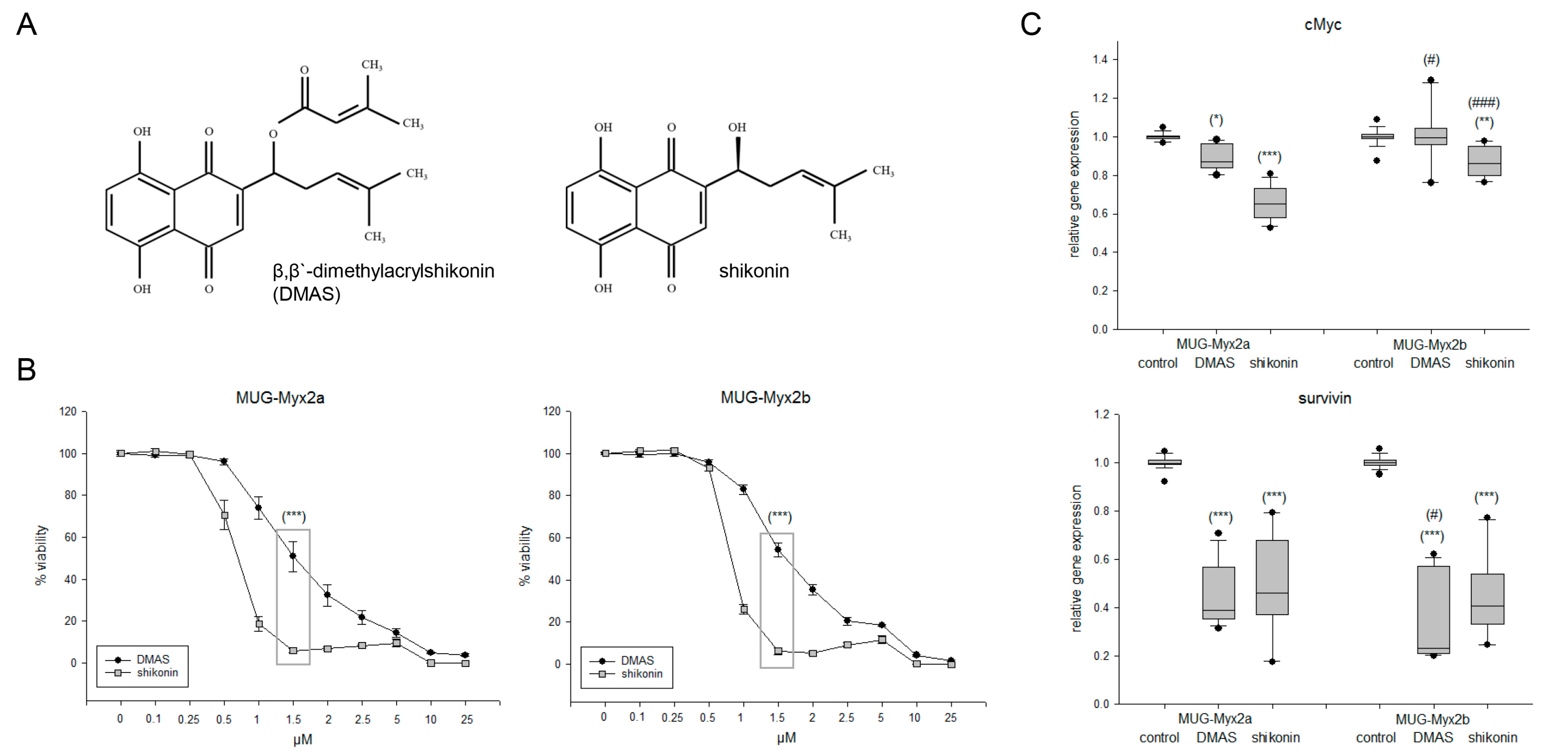
2.1. Effects of DMAS and Shikonin on Cell Viability and Proliferation
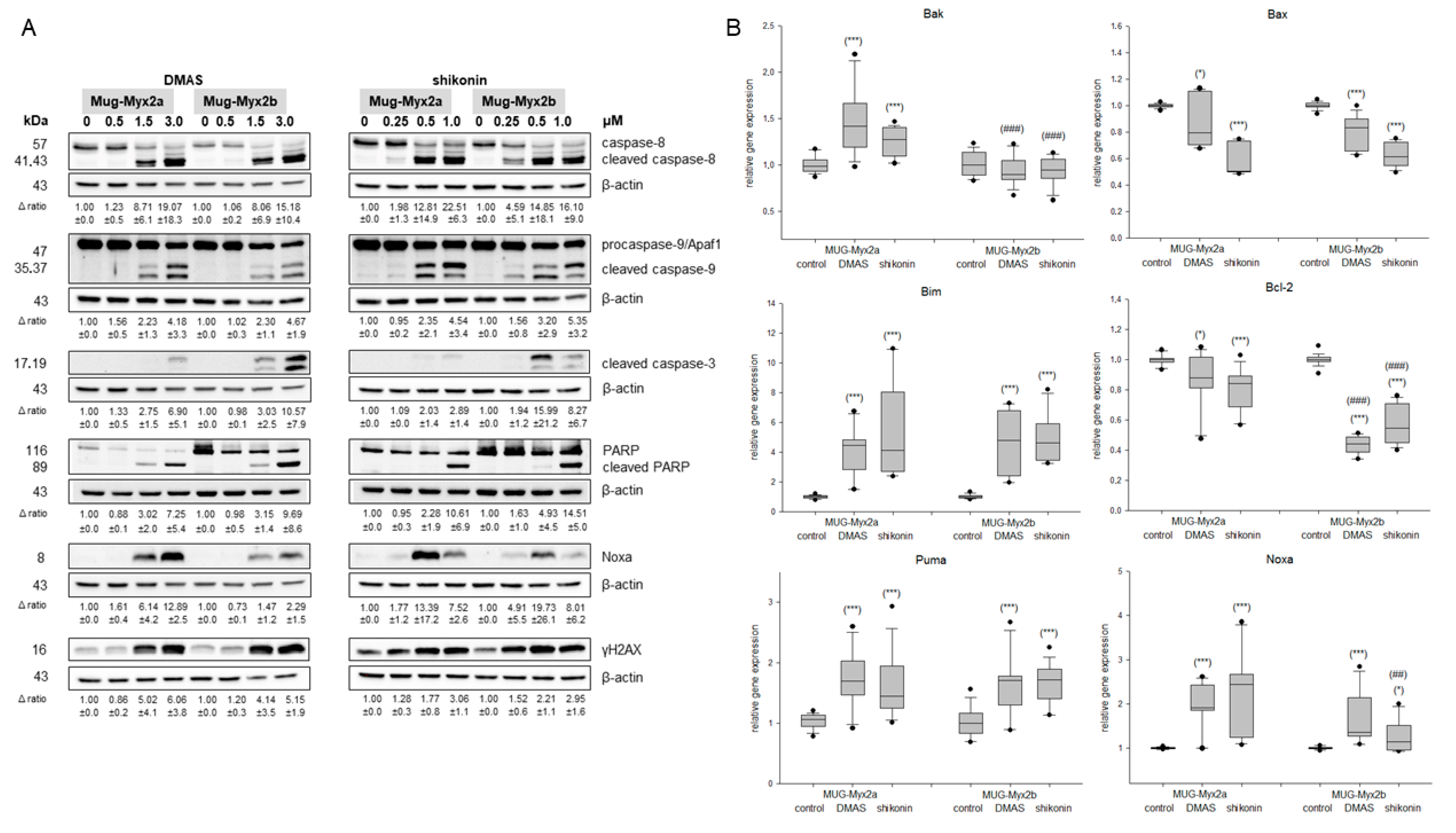
2.2. Caspase Activity and PARP Cleavage as Hallmarks of Apoptosis
2.3. The Expression of Death Receptors Was Influenced by Shikonin Derivatives
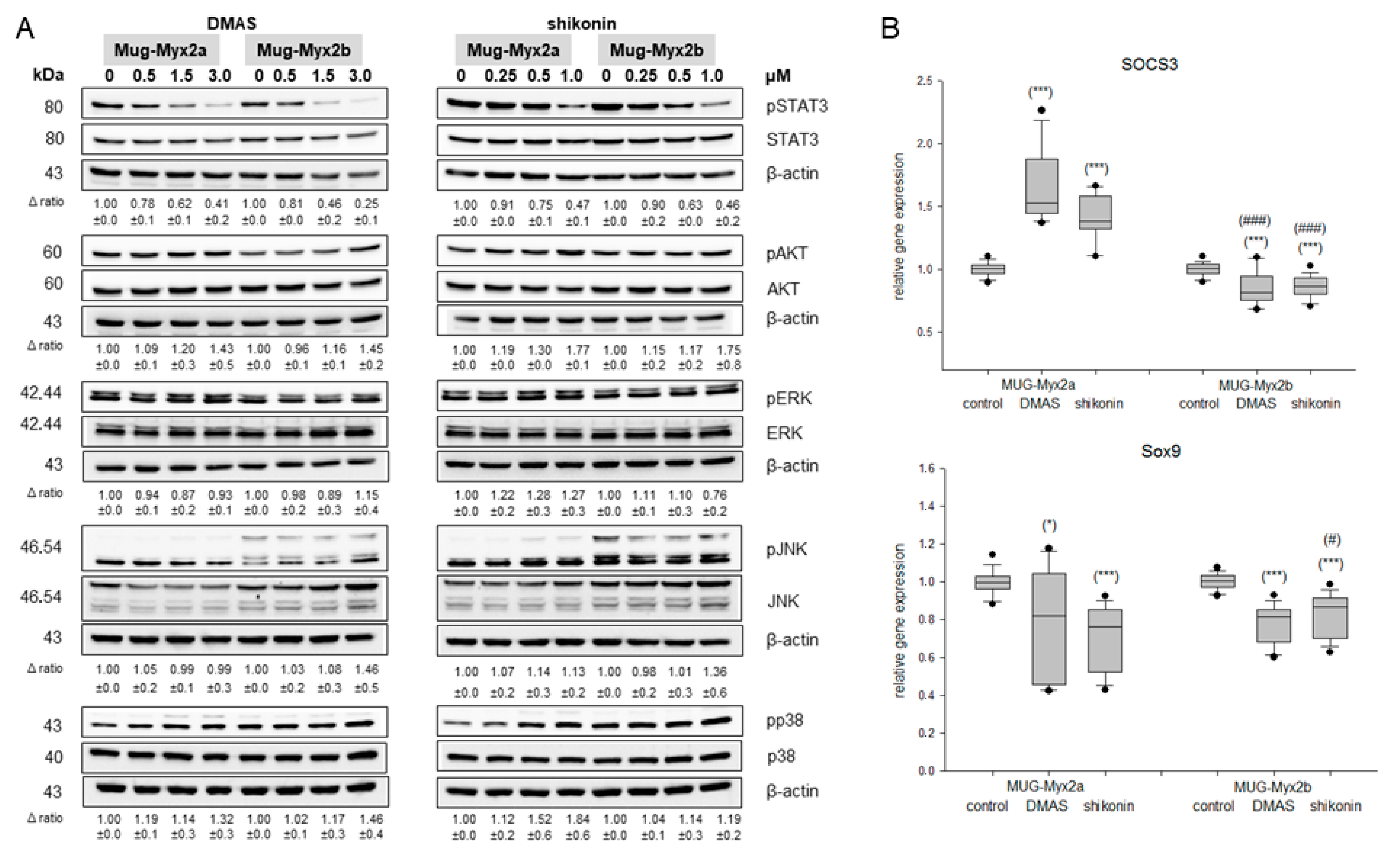
2.4. Modification of the Phosphorylation Pattern by DMAS and Shikonin
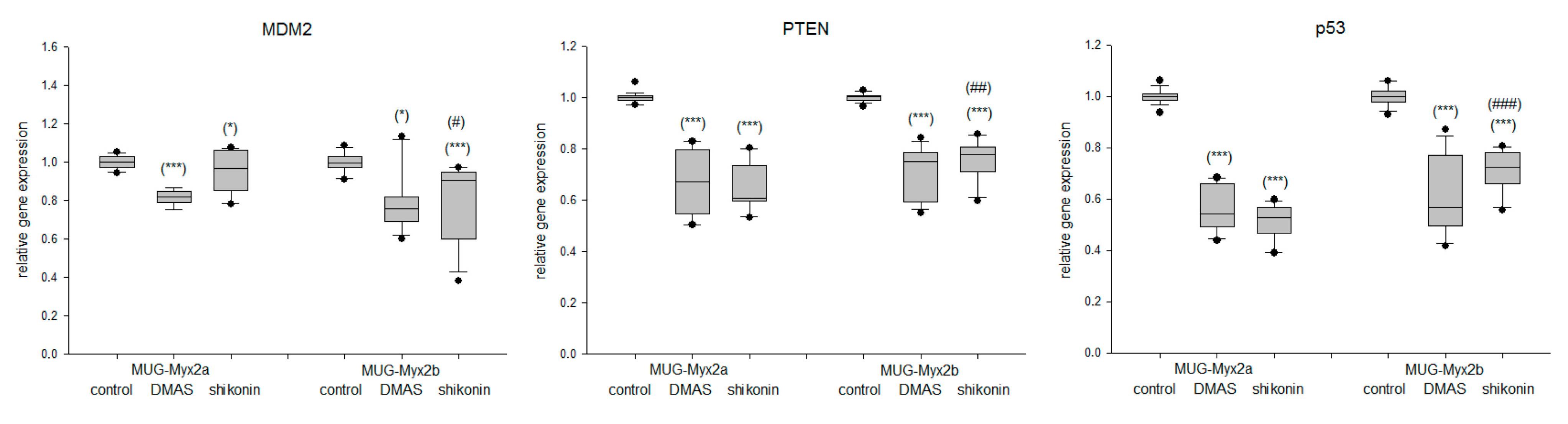
2.5. Effect of DMAS and Shikonin on DNA Damage Response
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Viability Assays
4.3. Western Blot Analysis
4.4. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nishio, J.; Iwasaki, H.; Nabeshima, K.; Naito, M. Cytogenetics and molecular genetics of myxoid soft-tissue sarcomas. Genet. Res. Int. 2011, 2011, 497148. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Mentzel, T.D.W.; Shibata, T.; World Health Organization (WHO). Classification of Soft Tissue and Bone Tumours, 5th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2020; pp. 124–126. [Google Scholar]
- Rhee, I.; Spazzoli, B.; Stevens, J.; Hansa, A.; Spelman, T.; Pang, G.; Guiney, M.; Powell, G.; Choong, P.; di Bella, C. Oncologic outcomes in myxofibrosarcomas: The role of a multidisciplinary approach and surgical resection margins. ANZ J. Surg. 2023, 93, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Boughzala-Bennadji, R.; Stoeckle, E.; le Péchoux, C.; Méeus, P.; Honoré, C.; Attal, J.; Duffaud, F.; de Pinieux, G.; Bompas, E.; Thariat, J.; et al. Localized myxofibrosarcomas: Roles of surgical margins and adjuvant radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 399–406. [Google Scholar] [CrossRef]
- Saxby, N.E.; An, Q.; Miller, B.J. Local recurrence of soft tissue sarcoma revisited: Is there a role for “selective” radiation? Iowa Orthop. J. 2022, 42, 239–248. [Google Scholar] [PubMed]
- Vanni, S.; De Vita, A.; Gurrieri, L.; Fausti, V.; Miserocchi, G.; Spadazzi, C.; Liverani, C.; Cocchi, C.; Calabrese, C.; Bongiovanni, A.; et al. Myxofibrosarcoma landscape: Diagnostic pitfalls, clinical management and future perspectives. Ther. Adv. Med. Oncol. 2022, 14, 17588359221093973. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; He, J.; Song, X.; Tan, L.; Wang, M.; Jiang, P.; Li, Y.; Cao, Z.; Peng, C. Pharmacological properties and derivatives of shikonin-A review in recent years. Pharmacol. Res. 2019, 149, 104463. [Google Scholar] [CrossRef]
- Boulos, J.C.; Rahama, M.; Hegazy, M.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef]
- Kretschmer, N.; Hufner, A.; Durchschein, C.; Popodi, K.; Rinner, B.; Lohberger, B.; Bauer, R. Naphthoquinones from Onosma paniculata induce cell-cycle arrest and apoptosis in melanoma cells. J. Nat. Prod. 2012, 75, 865–869. [Google Scholar] [CrossRef]
- Király, J.; Szabó, E.; Fodor, P.; Fejes, Z.; Nagy, B.; Juhász, É.; Vass, A.; Choudhury, M.; Kónya, G.; Halmos, G.; et al. Shikonin Causes an Apoptotic Effect on Human Kidney Cancer Cells through Ras/MAPK and PI3K/AKT Pathways. Molecules 2023, 28, 6725. [Google Scholar] [CrossRef]
- Deng, B.; Qiu, B. Shikonin inhibits invasiveness of osteosarcoma through MMP13 suppression. Tumour Biol. 2015, 36, 9311–9317. [Google Scholar] [CrossRef]
- Ni, F.; Huang, X.; Chen, Z.; Qian, W.; Tong, X. Shikonin exerts antitumor activity in Burkitt’s lymphoma by inhibiting C-MYC and PI3K/AKT/mTOR pathway and acts synergistically with doxorubicin. Sci. Rep. 2018, 8, 3317. [Google Scholar] [CrossRef] [PubMed]
- Lohberger, B.; Glänzer, D.; Kaltenegger, H.; Eck, N.; Leithner, A.; Bauer, R.; Kretschmer, N.; Steinecker-Frohnwieser, B. Shikonin derivatives cause apoptosis and cell cycle arrest in human chondrosarcoma cells via death receptors and MAPK regulation. BMC Cancer 2022, 22, 758. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, C.; Zhou, F.; Zhang, Y. Shikonin potentiates therapeutic efficacy of oxaliplatin through reactive oxygen species-mediated intrinsic apoptosis and endoplasmic reticulum stress in oxaliplatin-resistant colorectal cancer cells. Drug Dev. Res. 2023, 84, 542–555. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, H.; Wang, Z.; Li, H.; Gu, H.; Xia, E.; Yan, C.; Dai, Y.; Liu, C.; Wang, X.; et al. β, β-Dimethylacrylshikonin potentiates paclitaxel activity, suppresses immune evasion and triple negative breast cancer progression via STAT3Y705 phosphorylation inhibition based on network pharmacology and transcriptomics analysis. Phytomedicine 2023, 114, 154769. [Google Scholar] [CrossRef]
- Lohberger, B.; Stuendl, N.; Leithner, A.; Rinner, B.; Sauer, S.; Kashofer, K.; Liegl-Atzwanger, B. Establishment of a novel cellular model for myxofibrosarcoma heterogeneity. Sci. Rep. 2017, 7, 44700. [Google Scholar] [CrossRef]
- Pavlik, E.J.; Kenady, D.E.; van Nagell, J.R., Jr.; Hanson, M.B.; Donaldson, E.S.; Casper, S.; Garrett, D.; Smith, D.; Keaton, K.; Flanigan, R.C. Stability of doxorubicin in relation to chemosensitivity determinations: Loss of lethality and retention of antiproliferative activity. Cancer Investig. 1984, 2, 449–458. [Google Scholar] [CrossRef]
- Kretschmer, N.; Hufner, A.; Durchschein, C.; Popodi, K.; Rinner, B.; Lohberger, B.; Bauer, R. Synthesis and Pharmacological In Vitro Investigations of Novel Shikonin Derivatives with a Special Focus on Cyclopropane Bearing Derivatives. Int. J. Mol. Sci. 2021, 22, 2774. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Qi, H.; Zhang, X.; Liu, H.; Han, M.; Tang, X.; Qu, S.; Wang, X.; Yang, Y. Shikonin induced Apoptosis Mediated by Endoplasmic Reticulum Stress in Colorectal Cancer Cells. J. Cancer 2022, 13, 243–252. [Google Scholar] [CrossRef]
- Liu, C.; Xuan, L.Q.; Li, K.; Feng, Z.; Lv, C.; Li, X.J.; Ji, X.D.; Wan, R.; Shen, J. Shikonin Inhibits Cholangiocarcinoma Cell Line QBC939 by Regulating Apoptosis, Proliferation, and Invasion. Cell Transplant. 2021, 30, 963689720979162. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin and IAP proteins in cell-death mechanisms. Biochem. J. 2010, 430, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 3, 305–309. [Google Scholar]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, S.H.; Kim, Y.M.; Kim, S.H.; Yoo, E.S.; Woo, J.S.; Jung, G.H.; Jung, S.H.; Kim, B.S.; Jung, J.Y. Shikonin inhibits proliferation of melanoma cells by MAPK pathway-mediated induction of apoptosis. Biosci. Rep. 2021, 41, BSR20203834. [Google Scholar] [CrossRef]
- Guo, Z.L.; Li, J.Z.; Ma, Y.Y.; Qian, D.; Zhong, J.Y.; Jin, M.M.; Huang, P.; Che, L.Y.; Pan, B.; Wang, Y.; et al. Shikonin sensitizes A549 cells to TRAIL-induced apoptosis through the JNK, STAT3 and AKT pathways. BMC Cell Biol. 2018, 19, 29. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem. Sci. 2002, 27, 462–467. [Google Scholar] [CrossRef]
- Wang, F.; Jin, S.; Mayca Pozo, F.; Tian, D.; Tang, X.; Dai, Y.; Yao, X.; Tang, J.; Zhang, Y. Chemical screen identifies shikonin as a broad DNA damage response inhibitor that enhances chemotherapy through inhibiting ATM and ATR. Acta Pharm. Sin. B 2022, 12, 1339–1350. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Company; Order Number | Host Species | Clonality | Dilution | Incubation Time |
|---|---|---|---|---|---|
| cCaspase 8 | Santa Cruz; Sc-81656 | mouse | monoclonal | 1:1000 | ON 4 °C |
| cCaspase 9 | Santa Cruz; Sc-56076 | mouse | monoclonal | 1:1000 | ON 4 °C |
| cCaspase 3 | Cell signaling; CS, 9661 | rabbit | polyclonal | 1:1000 | ON 4 °C |
| cPARP | ThermoFisher; 436400 | mouse | monoclonal | 1:1000 | ON 4 °C |
| Noxa | ThermoFisher; MA1-41000 | mouse | monoclonal | 1:1000 | ON 4 °C |
| STAT3 | Cell signaling; CS, 4904 | rabbit | monoclonal | 1:1000 | ON 4 °C |
| pSTAT3 | Cell signaling; CS, 9145 | rabbit | monoclonal | 1:1000 | ON 4 °C |
| AKT | Cell signaling; CS, 9272 | rabbit | polyclonal | 1:1000 | ON 4 °C |
| pAKT | Cell signaling; CS, 9271 | rabbit | polyclonal | 1:1000 | ON 4 °C |
| ERK | Cell signaling; CS, 4695 | rabbit | monoclonal | 1:1000 | ON 4 °C |
| pERK | Cell signaling; CS, 4370 | rabbit | monoclonal | 1:1000 | ON 4 °C |
| JNK | Cell signaling; CS, 9252 | rabbit | polyclonal | 1:1000 | ON 4 °C |
| pJNK | Cell signaling; CS, 9251 | rabbit | polyclonal | 1:1000 | ON 4 °C |
| p38 | Cell signaling; CS, 9212 | rabbit | polyclonal | 1:1000 | ON 4 °C |
| pp38 | Cell signaling; CS, 4511 | rabbit | monoclonal | 1:1000 | ON 4 °C |
| pATR | Cell signaling; CS, 2853 | rabbit | polyclonal | 1:1000 | ON 4 °C |
| pATM | Cell signaling; CS, 13050 | rabbit | monoclonal | 1:1000 | ON 4 °C |
| MSH3 | Santa Cruz; Sc-271080 | mouse | monoclonal | 1:1000 | ON 4 °C |
| XPC | Santa Cruz; Sc-74410 | mouse | monoclonal | 1:1000 | ON 4 °C |
| pChk1 | Cell signaling; CS, 2348 | rabbit | monoclonal | 1:1000 | ON 4 °C |
| pCHk2 | Cell signaling; CS, 2197 | rabbit | monoclonal | 1:1000 | ON 4 °C |
| β-actin | Santa Cruz; Sc- 47778 | mouse | monoclonal | 1:15,000 | ON 4 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lohberger, B.; Kaltenegger, H.; Eck, N.; Glänzer, D.; Leithner, A.; Kretschmer, N. The Biological Assessment of Shikonin and β,β-dimethylacrylshikonin Using a Cellular Myxofibrosarcoma Tumor Heterogeneity Model. Int. J. Mol. Sci. 2023, 24, 15910. https://doi.org/10.3390/ijms242115910
Lohberger B, Kaltenegger H, Eck N, Glänzer D, Leithner A, Kretschmer N. The Biological Assessment of Shikonin and β,β-dimethylacrylshikonin Using a Cellular Myxofibrosarcoma Tumor Heterogeneity Model. International Journal of Molecular Sciences. 2023; 24(21):15910. https://doi.org/10.3390/ijms242115910
Chicago/Turabian StyleLohberger, Birgit, Heike Kaltenegger, Nicole Eck, Dietmar Glänzer, Andreas Leithner, and Nadine Kretschmer. 2023. "The Biological Assessment of Shikonin and β,β-dimethylacrylshikonin Using a Cellular Myxofibrosarcoma Tumor Heterogeneity Model" International Journal of Molecular Sciences 24, no. 21: 15910. https://doi.org/10.3390/ijms242115910
APA StyleLohberger, B., Kaltenegger, H., Eck, N., Glänzer, D., Leithner, A., & Kretschmer, N. (2023). The Biological Assessment of Shikonin and β,β-dimethylacrylshikonin Using a Cellular Myxofibrosarcoma Tumor Heterogeneity Model. International Journal of Molecular Sciences, 24(21), 15910. https://doi.org/10.3390/ijms242115910