
Metastatic ER+ Breast Cancer: Mechanisms of Resistance and Future Therapeutic Approaches

 , ,
, ,
Abstract
:1. Introduction
2. Endocrine Resistance
2.1. Mechanisms of Primary Endocrine Resistance
2.2. Mechanisms of Secondary Endocrine Resistance
2.2.1. ESR1 Mutations
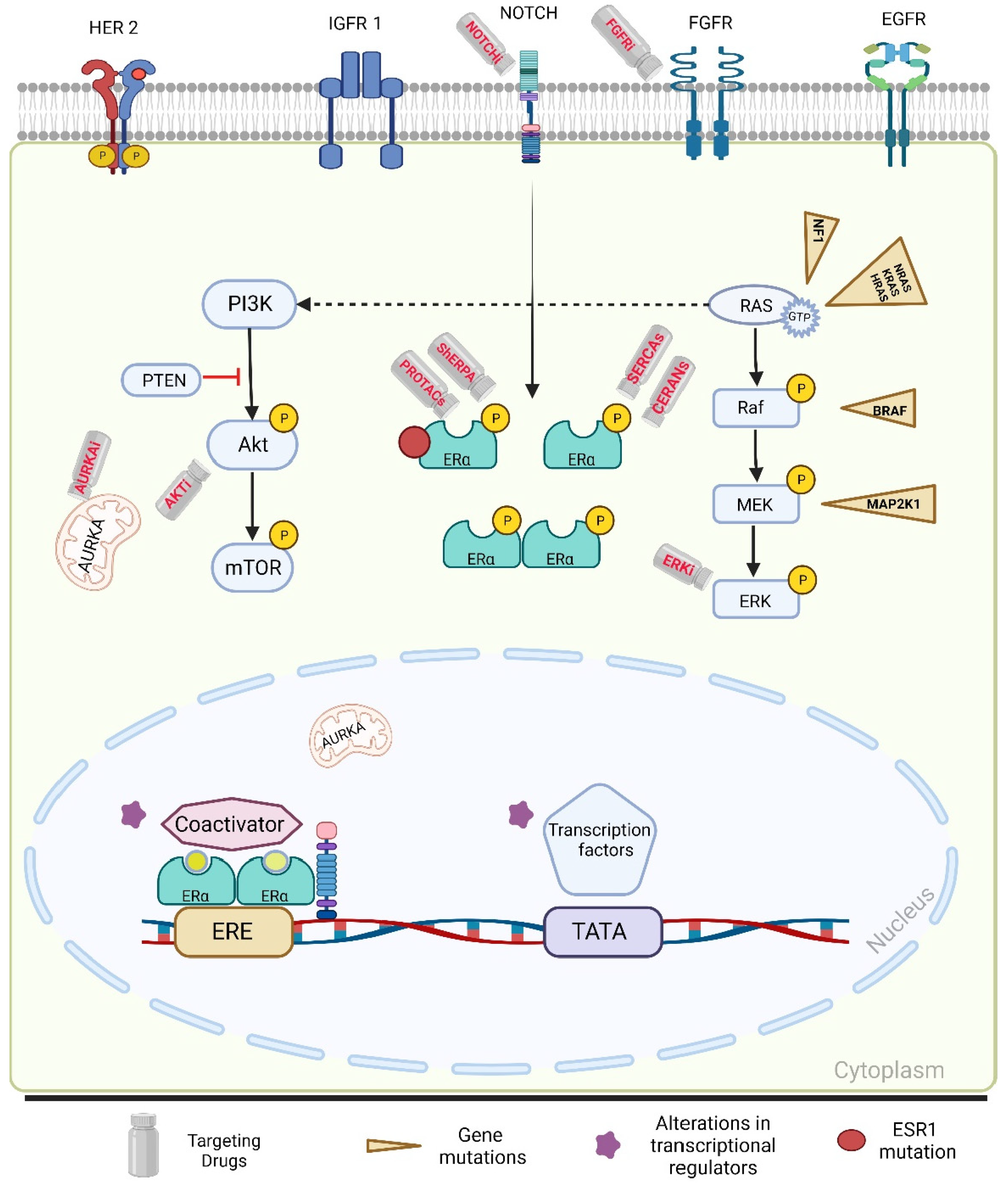
2.2.2. PI3K-Akt-mTORC1 Pathway
2.2.3. Mitogen-Activated Protein Kinase (MAPK) Pathway Alterations
2.2.4. Tumor Microenvironment
3. Current and Novel Therapeutic Approaches
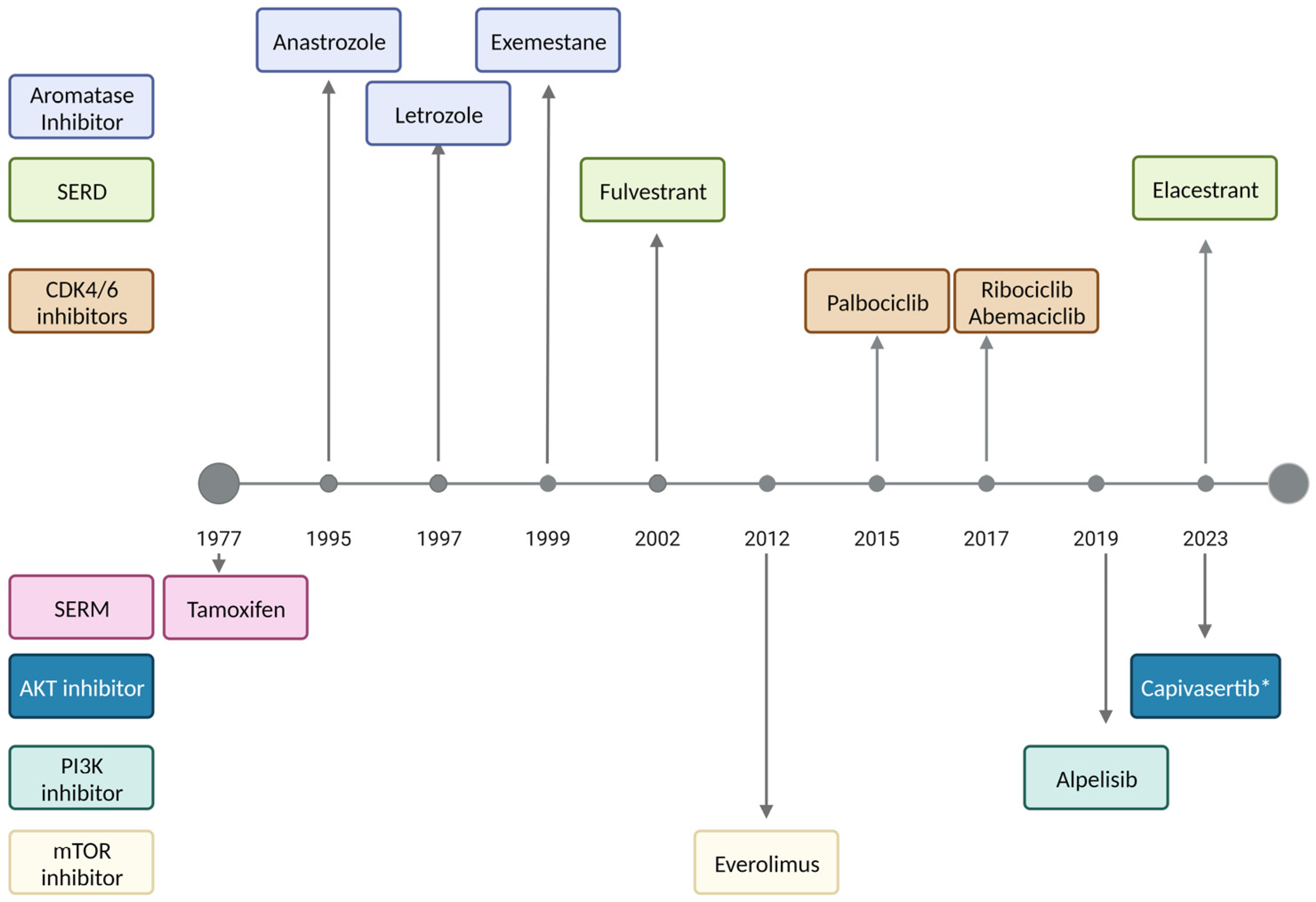
3.1. Antiestrogen Therapy
3.2. CDK4/6 Inhibitors
3.3. Everolimus
3.4. Alpelisib
3.5. Novel Therapies

{kind=link}
{kind=link}
| Mechanism of Action | Investigational Agents | |
|---|---|---|
| Oral SERDs [80,90,91,92] | Selective ER degraders | Camizestrant Imlunestrant Giredestrant |
| Novel SERMs [69,81,93] | Selective ER modulators | Lasofoxifene |
| SERM/SERD hybrids [94] | SERM/SERD hybrid | Basedoxifene |
| ShERPA [95,96] | Selective human ER partial agonist | TT-352 BMI-135 |
| PROTACs [97] | Proteolysis targeting chimera ER protein degrader | Vepdegestrant (ARV-471) |
| CERANs [83,84,98] | Complete estrogen receptor antagonist | Palazestrant (OP-1250) |
| ShERPA [68,99,100] | Selective estrogen receptor covalent antagonist | H3B-6545 |
| Antiprogestin [101] | Type I antiprogestin | Onapristone |
| AKT inhibitor [79,102] | Pan Akt kinase inhibitor | Capivasertib |
| ERK (MAPK) inhibitor [103,104] | Selective, ATP-competitive inhibitor of p38 MAPK α/β | Ralimetinib |
| FGFR inhibitor [105] | FGFR1–4 inhibitor | Futibatinib |
| Notch inhibitor [106] | Protein–protein interaction inhibitor that interferes with Notch transcription and signaling | CB-103 |
| AURKA inhibitor [107] | Selective Aurora A kinase inhibitor inducing apoptosis and autophagy | Alisertib |
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Clusan, L.; Ferrière, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, A.D.; Grischke, E.-M.; Brucker, S.Y. Endocrine-resistant breast cancer: Mechanisms and treatment. Breast Care 2020, 15, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Costa, A.; Cardoso, M.J.; Cufer, T.; El Saghir, N.; Fallowfield, L.; Fenech, D.; Francis, P.; Gelmon, K.; Giordano, S.H.; et al. ESO-ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2). Breast 2014, 23, 489–502. [Google Scholar] [CrossRef]
- D’souza, A.; Spicer, D.; Lu, J. Overcoming endocrine resistance in metastatic hormone receptor-positive breast cancer. J. Hematol. Oncol. 2018, 11, 80. [Google Scholar] [CrossRef]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Lopez, B.A.; Barrios, C.H.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)dagger. Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef]
- Bedard, P.L.; Freedman, O.C.; Howell, A.; Clemons, M. Overcoming endocrine resistance in breast cancer: Are signal transduction inhibitors the answer? Breast Cancer Res. Treat. 2008, 108, 307–317. [Google Scholar] [CrossRef]
- Stoner, M.; Saville, B.; Wormke, M.; Dean, D.; Burghardt, R.; Safe, S. Hypoxia Induces Proteasome-Dependent Degradation of Estrogen Receptor α in ZR-75 Breast Cancer Cells. Mol. Endocrinol. 2002, 16, 2231–2242. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Brett, J.O.; Spring, L.M.; Bardia, A.; Wander, S.A. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. 2021, 23, 85. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Fribbens, C.; O’leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Siddiqui, B.; Hu, J.; Pun, M.; Cornwell, M.; Buchwalter, G.; Hughes, M.E.; Wagle, N.; Kirschmeier, P.; Jänne, P.A.; et al. Unraveling the clinicopathological features driving the emergence of ESR1 mutations in metastatic breast cancer. npj Breast Cancer 2018, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, G.; Ma, C.X. Clinical Challenges in the Management of Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer: A Literature Review. Adv. Ther. 2020, 38, 109–136. [Google Scholar] [CrossRef]
- Turner, N.C.; Swift, C.; Kilburn, L.S.; Fribbens, C.; Beaney, M.; Garcia-Murillas, I.; Budzar, A.U.; Robertson, J.F.; Gradishar, W.; Piccart, M.; et al. ESR1 Mutations and Overall Survival on Fulvestrant versus Exemestane in Advanced Hormone Receptor–Positive Breast Cancer: A Combined Analysis of the Phase III SoFEA and EFECT Trials. Clin. Cancer Res. 2020, 26, 5172–5177. [Google Scholar] [CrossRef]
- Fribbens, C.; Murillas, I.G.; Beaney, M.; Hrebien, S.; O’leary, B.; Kilburn, L.; Howarth, K.; Epstein, M.; Green, E.; Rosenfeld, N.; et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann. Oncol. 2018, 29, 145–153. [Google Scholar] [CrossRef]
- Clatot, F.; Perdrix, A.; Beaussire, L.; Lequesne, J.; Lévy, C.; Emile, G.; Bubenheim, M.; Lacaille, S.; Calbrix, C.; Augusto, L.; et al. Risk of early progression according to circulating ESR1 mutation, CA-15.3 and cfDNA increases under first-line anti-aromatase treatment in metastatic breast cancer. Breast Cancer Res. 2020, 22, 56. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; De Angelis, C.; Brown, M.; Schiff, R. The Evolving Role of the Estrogen Receptor Mutations in Endocrine Therapy-Resistant Breast Cancer. Curr. Oncol. Rep. 2017, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Najim, O.; Seghers, S.; Sergoynne, L.; Van Gaver, H.; Papadimitriou, K.; Wouters, K.; Trinh, X.B.; Huizing, M.T.; Tjalma, W. The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: A systematic review and meta-analysis of randomized and non-randomized trials. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188315. [Google Scholar] [CrossRef]
- De Santo, I.; McCartney, A.; Migliaccio, I.; Di Leo, A.; Malorni, L. The Emerging Role of ESR1 Mutations in Luminal Breast Cancer as a Prognostic and Predictive Biomarker of Response to Endocrine Therapy. Cancers 2019, 11, 1894. [Google Scholar] [CrossRef]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-Therapy-Resistant ESR1 Variants Revealed by Genomic Characterization of Breast-Cancer-Derived Xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Hartmaier, R.; Trabucco, S.; Priedigkeit, N.; Chung, J.; Parachoniak, C.; Borre, P.V.; Morley, S.; Rosenzweig, M.; Gay, L.; Goldberg, M.; et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Ann. Oncol. 2018, 29, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gómez, H.; et al. Emergence of Constitutively Active Estrogen Receptor-α Mutations in Pretreated Advanced Estrogen Receptor–Positive Breast Cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef]
- Sermonix Pharmaceuticals Inc.; Linical Accelovance Group. Evaluation of Lasofoxifene Versus Fulvestrant in Advanced or Metastatic ER+/HER2- Breast Cancer with an ESR1 Mutation. 2019. Available online: https://classic.clinicaltrials.gov/show/NCT03781063 (accessed on 31 August 2023).
- Sermonix Pharmaceuticals Inc. Evaluation of Lasofoxifene Combined with Abemaciclib in Advanced or Metastatic ER+/HER2- Breast Cancer with an ESR1 Mutation. 2020. Available online: https://classic.clinicaltrials.gov/show/NCT04432454 (accessed on 31 August 2023).
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Millis, S.Z.; Ikeda, S.; Reddy, S.; Gatalica, Z.; Kurzrock, R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors. JAMA Oncol. 2016, 2, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Nunnery, S.E.; Mayer, I.A. Management of toxicity to isoform alpha-specific PI3K inhibitors. Ann. Oncol. 2019, 30, x21–x26. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V. Development of PI3K/AKT/mTOR Pathway Inhibitors and Their Application in Personalized Therapy for Non–Small-Cell Lung Cancer. J. Thorac. Oncol. 2012, 7, 1315–1326. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Kurokawa, H.; Lenferink, A.E.; Simpson, J.F.; Pisacane, P.I.; Sliwkowski, M.X.; Forbes, J.T.; Arteaga, C.L. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000, 60, 5887–5894. [Google Scholar]
- Stoica, G.E.; Franke, T.F.; Moroni, M.; Mueller, S.; Morgan, E.; Iann, M.C.; Winder, A.D.; Reiter, R.; Wellstein, A.; Martin, M.B.; et al. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene 2003, 22, 7998–8011. [Google Scholar] [CrossRef]
- Sanchez, C.G.; Ma, C.X.; Crowder, R.J.; Guintoli, T.; Phommaly, C.; Gao, F.; Lin, L.; Ellis, M.J. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011, 13, R21. [Google Scholar] [CrossRef]
- Miller, T.W.; Hennessy, B.T.; González-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; García-Echeverría, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor–positive human breast cancer. J. Clin. Investig. 2010, 120, 2406–2413. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, V.S.; Wang, H.; Nuovo, G.J.; Malbon, C.C. Hyperexpression of mitogen-activated protein kinase in human breast cancer. J. Clin. Investig. 1997, 99, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- von Lintig, F.C.; Dreilinger, A.D.; Varki, N.M.; Wallace, A.M.; Casteel, D.E.; Boss, G.R. Ras activation in human breast cancer. Breast Cancer Res. Treat. 2000, 62, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Mueller, H.; Flury, N.; Eppenberger-Castori, S.; Kueng, W.; Eppenberger, U.; David, F. Potential prognostic value of mitogen-activated protein kinase activity for disease-free survival of primary breast cancer patients. Int. J. Cancer 2000, 89, 384–388. [Google Scholar] [CrossRef]
- Pearson, A.; Proszek, P.Z.; Pascual, J.; Fribbens, C.; Shamsher, M.K.; Kingston, B.; O’Leary, B.; Herrera-Abreu, M.T.; Cutts, R.J.; Garcia-Murillas, I.; et al. Inactivating NF1 Mutations Are Enriched in Advanced Breast Cancer and Contribute to Endocrine Therapy Resistance. Clin. Cancer Res. 2020, 26, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.-L.; Baban, D.; et al. Estrogen receptor-α directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef]
- Morotti, M.; Bridges, E.; Valli, A.; Choudhry, H.; Sheldon, H.; Wigfield, S.; Gray, N.; Zois, C.E.; Grimm, F.; Jones, D.; et al. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 12452–12461. [Google Scholar] [CrossRef]
- Keller, S.; Mandl, I. Isolation of two peptides from acid hydrolyzates of elastin. Biochem. Biophys. Res. Commun. 1969, 35, 687–693. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Domchek, S.M.; Clark, A.S. Immunotherapy for Breast Cancer: What Are We Missing? Clin. Cancer Res. 2017, 23, 2640–2646. [Google Scholar] [CrossRef]
- Gibson, L.; Lawrence, D.; Dawson, C.; Bliss, J. Aromatase inhibitors for treatment of advanced breast cancer in postmenopausal women. Cochrane Database Syst. Rev. 2009, 2009, CD003370. [Google Scholar] [CrossRef]
- Bevers, T.B.; Niell, B.L.; Baker, J.L.; Bennett, D.L.; Bonaccio, E.; Camp, M.S.; Chikarmane, S.; Conant, E.F.; Eghtedari, M.; Flanagan, M.R.; et al. NCCN Guidelines(R) Insights: Breast Cancer Screening and Diagnosis, Version 1.2023. J. Natl. Compr. Canc Netw. 2023, 21, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.-C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.-A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results from the Randomized Phase III EMERALD Trial. J. Clin. Oncol. 2022, 40, 3246–3256. [Google Scholar] [CrossRef]
- Ibrance (palbociclib) Tablets [Prescribing Information]; Pfizer Labs: New York, NY, USA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/207103s008lbl.pdf (accessed on 30 May 2023).
- Kisqali (Ribociclib) Tablets; Prescribing Information; Novartis: East Hanover, NJ, USA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/209092s013,209935s021lbl.pdf (accessed on 15 September 2023).
- Verzenio (Abemaciclib) Tablets [Prescribing Information]; Lilly USA, LLC.: Indianapolis, IN, USA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/209092s013,209935s021lbl.pdf (accessed on 15 September 2023).
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2018, 29, 1541–1547. [Google Scholar] [CrossRef]
- Tripathy, D.; Im, S.-A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.-A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef]
- Wardell, S.E.; Ellis, M.J.; Alley, H.M.; Eisele, K.; VanArsdale, T.; Dann, S.G.; Arndt, K.T.; Primeau, T.; Griffin, E.; Shao, J.; et al. Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer. Clin. Cancer Res. 2015, 21, 5121–5130. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(-) Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Cortes, J.; Jerusalem, G.; Kaufman, P.A.; Ozyilkan, O.; Hegg, R.; Litchfield, L.M.; Wang, H.; Graham, H.T.; Wijayawardana, S.R.; et al. Abstract 785: Genomic markers of response to monotherapy abemaciclib in the nextMONARCH 1 study. Cancer Res. 2020, 80, 785. [Google Scholar] [CrossRef]
- Goetz, M.P.; Hamilton, E.P.; Campone, M.; Hurvitz, S.A.; Cortes, J.; Johnston, S.R.D.; Jerusalem, G.H.M.; Graham, H.; Wang, H.; Litchfield, L.; et al. Acquired genomic alterations in circulating tumor DNA from patients receiving abemaciclib alone or in combination with endocrine therapy. J. Clin. Oncol. 2020, 38, 3519. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., III; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Noguchi, S.; Pritchard, K.I.; Burris, H.A., III; Baselga, J.; Gnant, M.; Hortobagyi, G.N.; Campone, M.; Pistilli, B.; Piccart, M.; et al. Everolimus Plus Exemestane in Postmenopausal Patients with HR+ Breast Cancer: BOLERO-2 Final Progression-Free Survival Analysis. Adv. Ther. 2013, 30, 870–884. [Google Scholar] [CrossRef]
- Yamnik, R.L.; Holz, M.K. mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor α serine 167 phosphorylation. FEBS Lett. 2010, 584, 124–128. [Google Scholar] [CrossRef]
- Yamnik, R.L.; Digilova, A.; Davis, D.C.; Brodt, Z.; Murphy, C.J.; Holz, M.K. S6 Kinase 1 Regulates Estrogen Receptor α in Control of Breast Cancer Cell Proliferation. J. Biol. Chem. 2009, 284, 6361–6369. [Google Scholar] [CrossRef]
- Efeyan, A.; Sabatini, D.M. mTOR and cancer: Many loops in one pathway. Curr. Opin. Cell Biol. 2010, 22, 169–176. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N. Engl. J. Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef]
- Piqray (alpelisib) [Prescribing Information]; Novartis Pharmaceuticals Corporation: East Hanover, NJ, USA, 2022.
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018, 23, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Arciero, C.A.; Guo, Y.; Jiang, R.; Behera, M.; O’Regan, R.; Peng, L.; Li, X. ER+/HER2+ Breast Cancer Has Different Metastatic Patterns and Better Survival Than ER−/HER2+ Breast Cancer. Clin. Breast Cancer 2019, 19, 236–245. [Google Scholar] [CrossRef]
- AstraZeneca, Capivasertib+Fulvestrant vs. Placebo+Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic HR+/HER2- Breast Cancer. 2020. Available online: https://classic.clinicaltrials.gov/show/NCT04305496 (accessed on 30 August 2023).
- Oliveira, M.; Pominchuck, D.; Nowecki, Z.; Hamilton, E.; Kulyaba, Y.; Andabekov, T.; Hotko, Y.; Melkadze, T.; Nemsadze, G.; Neven, P.; et al. Abstract GS3-02: GS3-02 Camizestrant, a next generation oral SERD vs. fulvestrant in post-menopausal women with advanced ER-positive HER2-negative breast cancer: Results of the randomized, multi-dose Phase 2 SERENA-2 trial. Cancer Res. 2023, 83, GS3-02. [Google Scholar] [CrossRef]
- Lainé, M.; Fanning, S.W.; Chang, Y.-F.; Green, B.; Greene, M.E.; Komm, B.; Kurleto, J.D.; Phung, L.; Greene, G.L. Lasofoxifene as a potential treatment for therapy-resistant ER-positive metastatic breast cancer. Breast Cancer Res. 2021, 23, 54. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Sun, X.; Yang, Y.; Xia, L.; Wang, S.; Fu, Y.; Zhu, Y.; Xu, S.; Zhu, W. A Novel Dual PI3K/mTOR Inhibitor, XIN-10, for the Treatment of Cancer. Int. J. Mol. Sci. 2023, 24, 14821. [Google Scholar] [CrossRef]
- Olema Pharmaceuticals, I.; Pfizer, A. Phase 1 Study of Oral OP-1250 in Combination with Palbociclib in HR+/HER2- Breast Cancer Patients. 2021. Available online: https://classic.clinicaltrials.gov/show/NCT05266105 (accessed on 31 August 2023).
- Patel, R.; Klein, P.; Tiersten, A.; Sparano, J.A. An emerging generation of endocrine therapies in breast cancer: A clinical perspective. npj Breast Cancer 2023, 9, 20. [Google Scholar] [CrossRef]
- Xiong, R.; Patel, H.K.; Gutgesell, L.M.; Zhao, J.; Delgado-Rivera, L.; Pham, T.N.D.; Zhao, H.; Carlson, K.; Martin, T.; Katzenellenbogen, J.A.; et al. Selective Human Estrogen Receptor Partial Agonists (ShERPAs) for Tamoxifen-Resistant Breast Cancer. J. Med. Chem. 2016, 59, 219–237. [Google Scholar] [CrossRef]
- University of Wisconsin, Madison; Context Therapeutics Inc. Onapristone and Fulvestrant for ER+ HER2- Metastatic Breast Cancer after Endocrine Therapy and CDK4/6 Inhibitors (The SMILE Study). 2021. Available online: https://classic.clinicaltrials.gov/show/NCT04738292 (accessed on 30 August 2023).
- Kamaraju, S.; Fowler, A.M.; Weil, E.; Wisinski, K.B.; Truong, T.H.; Lehr, M.; Chaudhary, L.N.; Cheng, Y.C.; Chitambar, C.R.; Rui, H.; et al. Leveraging Antiprogestins in the Treatment of Metastatic Breast Cancer. Endocrinology 2021, 162. [Google Scholar] [CrossRef]
- Niu, H.; Manfredi, M.; Ecsedy, J.A. Scientific Rationale Supporting the Clinical Development Strategy for the Investigational Aurora A Kinase Inhibitor Alisertib in Cancer. Front. Oncol. 2015, 5, 189. [Google Scholar] [CrossRef]
- López-Mejía, J.A.; Mantilla-Ollarves, J.C.; Rocha-Zavaleta, L. Modulation of JAK-STAT Signaling by LNK: A Forgotten Oncogenic Pathway in Hormone Receptor-Positive Breast Cancer. Int. J. Mol. Sci. 2023, 24, 14777. [Google Scholar] [CrossRef]
- Oliveira, M.; Pominchuk, D.; Hamilton, E.P.; Kulyaba, Y.; Melkadze, T.; Neven, P.; Nemsadze, G.; Andabekov, T.; Semegen, Y.; Hotko, Y.; et al. Clinical activity of camizestrant, a next-generation SERD, versus fulvestrant in patients with a detectable ESR1 mutation: Exploratory analysis of the SERENA-2 phase 2 trial. J. Clin. Oncol. 2023, 41, 1066. [Google Scholar] [CrossRef]
- Jhaveri, K.; Wang, H.-C.; Ma, C.; Lim, E.; Tao, J.J.; Manso, L.; Pierga, J.-Y.; Parajuli, R.; Gilarranz, Y.J.; Lu, Y.-S.; et al. Abstract PD13-12: PD13-12 Imlunestrant, an oral selective estrogen receptor degrader, in combination with abemaciclib with or without an aromatase inhibitor, in estrogen receptor-positive advanced breast cancer: Results from the phase 1a/b EMBER study. Cancer Res. 2023, 83, PD13-12. [Google Scholar] [CrossRef]
- Mayer, E.L.; Tolaney, S.; Brufsky, A.M.; Gradishar, W.; Jhaveri, K.; Martín, M.; Moscetti, L.; Vidal, G.; Cortazar, P.; Feldman, M.; et al. Abstract OT2-01-07: evERA Breast Cancer: A phase III study of giredestrant (GDC-9545)+ everolimus vs. exemestane+ everolimus in patients with estrogen receptor+, HER2–locally advanced or metastatic breast cancer. Cancer Res. 2023, 83, OT2-01-07. [Google Scholar] [CrossRef]
- Damodaran, S.; Moore, H.C.; Anderson, I.C.; Cherian, M.A.; O’Sullivan, C.C.; Plourde, P.V.; Portman, D.J.; Goetz, M.P. Lasofoxifene (LAS) plus abemaciclib (Abema) for treating ESR1-mutated ER+/HER2-metastatic breast cancer (mBC) after progression on prior therapies: ELAINE 2 study update. J. Clin. Oncol. 2023, 41, 1057. [Google Scholar] [CrossRef]
- Fanning, S.W.; Jeselsohn, R.; Dharmarajan, V.; Mayne, C.G.; Karimi, M.; Buchwalter, G.; Houtman, R.; Toy, W.; Fowler, C.E.; Han, R.; et al. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. Elife 2018, 7, e37161. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.Z.; Liu, L.C.; Fischer, J.H.; Wiley, E.L.; Sachdev, J.C.; Bleeker, J.; Hurley, R.W.; Tonetti, D.A.; Thatcher, G.R.J.; Venuti, R.P.; et al. Phase 1 study of TTC-352 in patients with metastatic breast cancer progressing on endocrine and CDK4/6 inhibitor therapy. Breast Cancer Res. Treat. 2020, 183, 617–627. [Google Scholar] [CrossRef]
- Abderrahman, B.; Maximov, P.Y.; Curpan, R.F.; Hanspal, J.S.; Fan, P.; Xiong, R.; Tonetti, D.A.; Thatcher, G.R.; Jordan, V.C. Pharmacology and Molecular Mechanisms of Clinically Relevant Estrogen Estetrol and Estrogen Mimic BMI-135 for the Treatment of Endocrine-Resistant Breast Cancer. Mol. Pharmacol. 2020, 98, 364–381. [Google Scholar] [CrossRef]
- Schott, A.F.; Hurvitz, S.; Ma, C.; Hamilton, E.; Nanda, R.; Zahrah, G.; Hunter, N.; Tan, A.R.; Telli, M.; Mesias, J.A.; et al. Abstract GS3-03: GS3-03 ARV-471, a PROTAC® estrogen receptor (ER) degrader in advanced ER-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer: Phase 2 expansion (VERITAC) of a phase 1/2 study. Cancer Res. 2023, 83, GS3-03. [Google Scholar] [CrossRef]
- Buono, G.; Deleuze, A.; Klocker, E.; Nardin, S.; Eiriz, I.; Luz, P.; Braga, S.; Harbeck, N.; Dieras, V. 237P Real-world safety and efficacy of trastuzumab-deruxtecan (T-DXd) in HER2-positive advanced breast cancer (ABC) elderly patients (pts): The TREX-Old retrospective registry. ESMO Open 2023, 8. [Google Scholar] [CrossRef]
- Fasching, P.A.; Yadav, S.; Hu, C.; Wunderle, M.; Häberle, L.; Hart, S.N.; Rübner, M.; Polley, E.C.; Lee, K.Y.; Gnanaolivu, R.D.; et al. Mutations in BRCA1/2 and Other Panel Genes in Patients With Metastatic Breast Cancer -Association With Patient and Disease Characteristics and Effect on Prognosis. J. Clin. Oncol. 2021, 39, 1619–1630. [Google Scholar] [CrossRef]
- Hamilton, E.P.; Wang, J.S.; Pluard, T.J.; Johnston, S.R.D.; Morikawa, A.; Dees, E.C.; Jones, R.H.; Haley, B.B.; Armstrong, A.C.; Cohen, A.L.; et al. Phase I/II study of H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer. J. Clin. Oncol. 2021, 39, 1018. [Google Scholar] [CrossRef]
- Hamilton, E.; Pusztai, L.; Soliman, H.H.; Hurvitz, S.; Grzegorzewski, K.; Habboubi, N.; Marreddy, P.; Sahmoud, T.; Ibrahim, N. Abstract OT2-01-04: ELONA: An open-label, phase 1b-2 study of elacestrant, in combination with onapristone in patients with estrogen receptor-positive, progesterone receptor-positive, HER2-negative advanced or metastatic breast cancer. Cancer Res. 2023, 83, OT2-01. [Google Scholar] [CrossRef]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Moreno, H.L.G.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; et al. Capivasertib in Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2023, 388, 2058–2070. [Google Scholar] [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non–Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef]
- Vergote, I.; Heitz, F.; Buderath, P.; Powell, M.A.; Sehouli, J.; Lee, C.M.; Hamilton, A.L.; Fiorica, J.; Moore, K.N.; Teneriello, M.; et al. A randomized, double-blind, placebo-controlled phase Ib/II study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine (G) and carboplatin (C) versus GC for women with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2019, 37, 5537. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Bahleda, R.; Hierro, C.; Sanson, M.; Bridgewater, J.; Arkenau, H.-T.; Tran, B.; Kelley, R.K.; Park, J.O.; Javle, M.; et al. Futibatinib, an Irreversible FGFR1-4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov. 2022, 12, 402–415. [Google Scholar] [CrossRef]
- Vigolo, M.; Urech, C.; Lamy, S.; Monticone, G.; Zabaleta, J.; Hossain, F.; Wyczechowska, D.; Del Valle, L.; O’regan, R.M.; Miele, L.; et al. The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer. Cancers 2023, 15, 3957. [Google Scholar] [CrossRef]
- Haddad, T.C.; Suman, V.J.; D’Assoro, A.B.; Carter, J.M.; Giridhar, K.V.; McMenomy, B.P.; Santo, K.; Mayer, E.L.; Karuturi, M.S.; Morikawa, A.; et al. Evaluation of Alisertib Alone or Combined with Fulvestrant in Patients with Endocrine-Resistant Advanced Breast Cancer: The Phase 2 TBCRC041 Randomized Clinical Trial. JAMA Oncol. 2023, 9, 815–824. [Google Scholar] [CrossRef]
- Harris, L.N.; Blanke, C.D.; Erba, H.P.; Ford, J.M.; Gray, R.J.; LeBlanc, M.L.; Hu-Lieskovan, S.; Litzow, M.R.; Luger, S.M.; Meric-Bernstam, F.; et al. The New NCI Precision Medicine Trials. Clin. Cancer Res. 2023, OF1–OF5. [Google Scholar] [CrossRef]
- Batalini, F.; Gulhan, D.C.; Mao, V.; Tran, A.; Polak, M.; Xiong, N.; Tayob, N.; Tung, N.M.; Winer, E.P.; Mayer, E.L.; et al. Mutational Signature 3 Detected from Clinical Panel Sequencing is Associated with Responses to Olaparib in Breast and Ovarian Cancers. Clin. Cancer Res. 2022, 28, 4714–4723. [Google Scholar] [CrossRef]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Tivey, A.; Church, M.; Rothwell, D.; Dive, C.; Cook, N. Circulating tumour DNA—Looking beyond the blood. Nat. Rev. Clin. Oncol. 2022, 19, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, E.; Zhao, E.; Bratman, S.V.; Siu, L.L. Monitoring and adapting cancer treatment using circulating tumor DNA kinetics: Current research, opportunities, and challenges. Sci. Adv. 2022, 8, eabi8618. [Google Scholar] [CrossRef] [PubMed]
- Royal Marsden NHS Foundation Trust; Massachusetts General Hospital; Natera, Inc. Safe De-escalation of Chemotherapy for Stage 1 Breast Cancer. 2023. Available online: https://classic.clinicaltrials.gov/show/NCT05058183 (accessed on 24 September 2023).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raheem, F.; Karikalan, S.A.; Batalini, F.; El Masry, A.; Mina, L. Metastatic ER+ Breast Cancer: Mechanisms of Resistance and Future Therapeutic Approaches. Int. J. Mol. Sci. 2023, 24, 16198. https://doi.org/10.3390/ijms242216198
Raheem F, Karikalan SA, Batalini F, El Masry A, Mina L. Metastatic ER+ Breast Cancer: Mechanisms of Resistance and Future Therapeutic Approaches. International Journal of Molecular Sciences. 2023; 24(22):16198. https://doi.org/10.3390/ijms242216198
Chicago/Turabian StyleRaheem, Farah, Suganya Arunachalam Karikalan, Felipe Batalini, Aya El Masry, and Lida Mina. 2023. "Metastatic ER+ Breast Cancer: Mechanisms of Resistance and Future Therapeutic Approaches" International Journal of Molecular Sciences 24, no. 22: 16198. https://doi.org/10.3390/ijms242216198
APA StyleRaheem, F., Karikalan, S. A., Batalini, F., El Masry, A., & Mina, L. (2023). Metastatic ER+ Breast Cancer: Mechanisms of Resistance and Future Therapeutic Approaches. International Journal of Molecular Sciences, 24(22), 16198. https://doi.org/10.3390/ijms242216198