
Key Genes of the Immune System and Predisposition to Acquired Hemophilia A: Evidence from a Spanish Cohort of 49 Patients Using Next-Generation Sequencing
 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Results
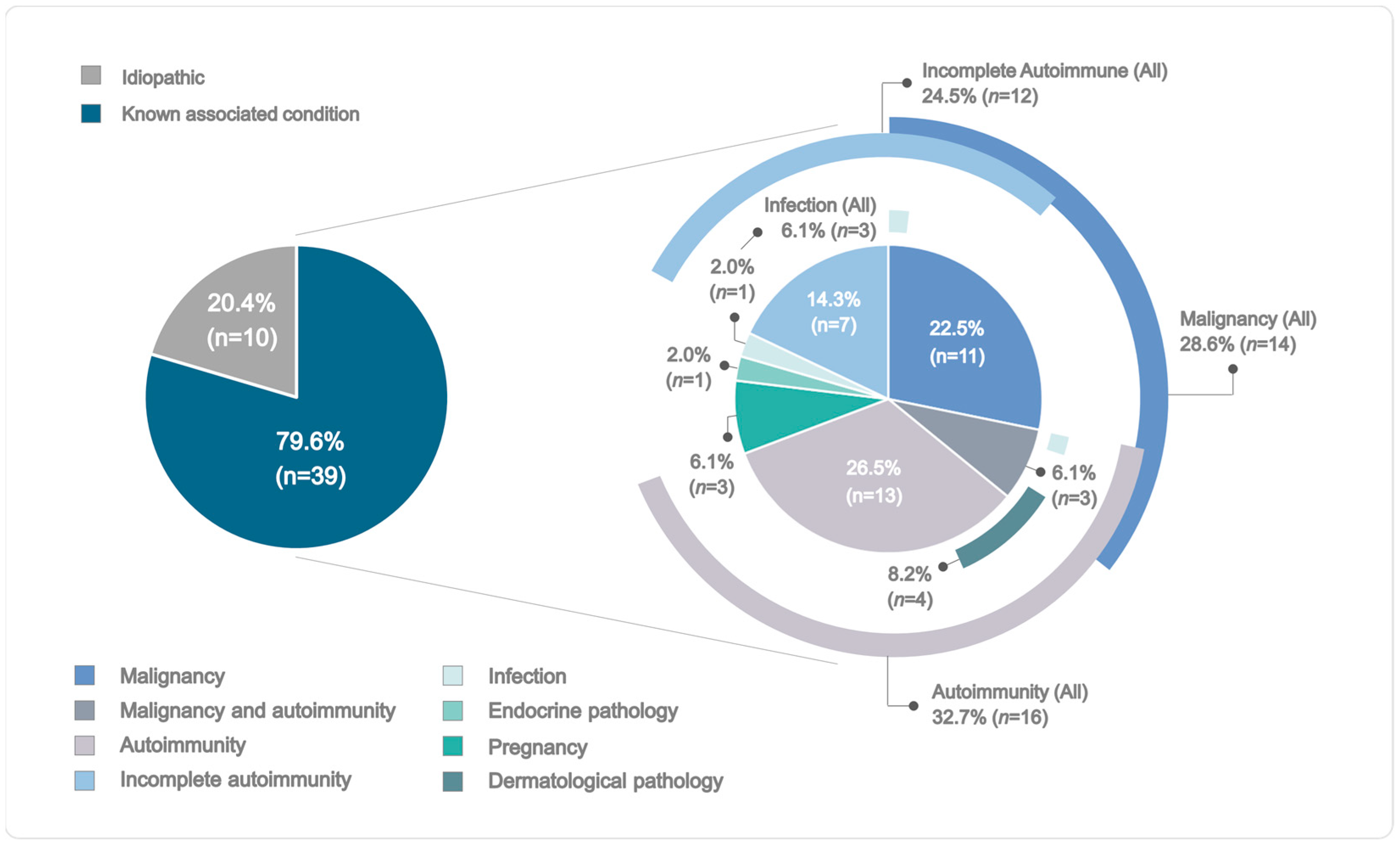
2.1. Demographic and Clinical Profiles of the AHA Cohort
2.2. Hemostatic Treatment and Immunosuppressive Therapy
2.3. Genetic Analyses and Association Studies
2.3.1. Classical HLA Class I and II Genes
2.3.2. Non-Classical HLA Class I and HLA Class I-like Genes
2.3.3. KIR Genes, Genotypes, and Haplotypes
2.3.4. KLRK1 Gene
3. Discussion
4. Methods
4.1. Patients
4.2. Sample Collection and Phenotype Determinations
4.3. Candidate Region Amplification and Sequencing
4.4. Bioinformatics Analyses
4.5. Reference Samples
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kruse-Jarres, R.; Kempton, C.L.; Baudo, F.; Collins, P.W.; Knoebl, P.; Leissinger, C.A.; Tiede, A.; Kessler, C.M. Acquired Hemophilia A: Updated Review of Evidence and Treatment Guidance. Am. J. Hematol. 2017, 92, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Gandini, G.; Di Paolantonio, T.; Mariani, G. Acquired Hemophilia A: A Concise Review. Am. J. Hematol. 2005, 80, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.W.; Hirsch, S.; Baglin, T.P.; Dolan, G.; Hanley, J.; Makris, M.; Keeling, D.M.; Liesner, R.; Brown, S.A.; Hay, C.R.M. Acquired Hemophilia A in the United Kingdom: A 2-Year National Surveillance Study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007, 109, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Tay, L.; Duncan, E.; Singhal, D.; Al-Qunfoidi, R.; Coghlan, D.; Jaksic, W.; Szabo, F.; McRae, S.; Lloyd, J. Twelve Years of Experience of Acquired Hemophilia A: Trials and Tribulations in South Australia. Semin. Thromb. Hemost. 2009, 35, 769–777. [Google Scholar] [CrossRef]
- Pardos-Gea, J.; Fernández-Díaz, N.; Parra, R.; Cortina, V.; Altisent, C. Diagnostic Delay in Acquired Haemophilia: Analysis of Causes and Consequences in a 20-Year Spanish Cohort. Haemophilia 2018, 24, e163–e166. [Google Scholar] [CrossRef]
- Tiede, A.; Collins, P.; Knoebl, P.; Teitel, J.; Kessler, C.; Shima, M.; Minno, G.D.; d’Oiron, R.; Salaj, P.; Jiménez-Yuste, V.; et al. International Recommendations on the Diagnosis and Treatment of Acquired Hemophilia A. Haematologica 2020, 105, 1791–1801. [Google Scholar] [CrossRef]
- Reding, M.T.; Wu, H.; Krampf, M.; Okita, D.K.; Diethelm-Okita, B.M.; Key, N.S.; Conti-Fine, B.M. CD4+ T Cell Response to Factor VIII in Hemophilia A, Acquired Hemophilia, and Healthy Subjects. Thromb. Haemost. 1999, 82, 509–515. [Google Scholar] [CrossRef]
- Reding, M.T.; Wu, H.; Krampf, M.; Okita, D.K.; Diethelm-Okita, B.M.; Christie, B.A.; Key, N.S.; Conti-Fine, B.M. Sensitization of CD4+ T Cells to Coagulation Factor VIII: Response in Congenital and Acquired Hemophilia Patients and in Healthy Subjects. Thromb. Haemost. 2000, 84, 643–652. [Google Scholar] [CrossRef]
- Reding, M.T.; Lei, S.; Lei, H.; Green, D.; Gill, J.; Conti-Fine, B.M. Distribution of Th1- and Th2-Induced Anti-Factor VIII IgG Subclasses in Congenital and Acquired Hemophilia Patients. Thromb. Haemost. 2002, 88, 568–575. [Google Scholar]
- Pavlova, A.; Diaz-Lacava, A.; Zeitler, H.; Satoguina, J.; Niemann, B.; Krause, M.; Scharrer, I.; Hoerauf, A.; Wienker, T.; Oldenburg, J. Increased Frequency of the CTLA-4 49 A/G Polymorphism in Patients with Acquired Haemophilia A Compared to Healthy Controls. Haemophilia 2008, 14, 355–360. [Google Scholar] [CrossRef]
- Tiede, A.; Eisert, R.; Czwalinna, A.; Miesbach, W.; Scharrer, I.; Ganser, A. Acquired Haemophilia Caused by Non-Haemophilic Factor VIII Gene Variants. Ann. Hematol. 2010, 89, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Lim, J.A.; Kim, H.C.; Lee, H.W.; Kim, H.S. Identification of a Shared F8 Mutation in the Korean Patients with Acquired Hemophilia A. Korean J. Hematol. 2011, 46, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.; Zeitler, H.; Scharrer, I.; Brackmann, H.H.; Oldenburg, J. HLA Genotype in Patients with Acquired Haemophilia A. Haemophilia 2010, 16, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Schleinitz, N.; Vély, F.; Harlé, J.R.; Vivier, E. Natural Killer Cells in Human Autoimmune Diseases. Immunology 2010, 131, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Cheent, K.; Khakoo, S.I. Natural Killer Cells: Integrating Diversity with Function. Immunology 2009, 126, 449–457. [Google Scholar] [CrossRef]
- Van Belle, T.L.; von Herrath, M.G. The Role of the Activating Receptor NKG2D in Autoimmunity. Mol. Immunol. 2009, 47, 8–11. [Google Scholar] [CrossRef]
- Hayashi, T.; Imai, K.; Morishita, Y.; Hayashi, I.; Kusunoki, Y.; Nakachi, K. Identification of the NKG2D Haplotypes Associated with Natural Cytotoxic Activity of Peripheral Blood Lymphocytes and Cancer Immunosurveillance. Cancer Res. 2006, 66, 563–570. [Google Scholar] [CrossRef]
- Gonzalez-Galarza, F.F.; McCabe, A.; Santos, E.J.M.D.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Cid-Pavon, G.M.D.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acids Res. 2020, 48, D783–D788. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Schep, S.J.; van Dijk, W.E.M.; Beckers, E.A.M.; Meijer, K.; Coppens, M.; Eikenboom, J.; Leebeek, F.W.G.; van Vulpen, L.F.D.; Fischer, K.F.; Schutgens, R.E.G. Treatment of Acquired Hemophilia A, a Balancing Act: Results from a 27-Year Dutch Cohort Study. Am. J. Hematol. 2021, 96, 51–59. [Google Scholar] [CrossRef]
- Tiede, A.; Klamroth, R.; Scharf, R.E.; Trappe, R.U.; Holstein, K.; Huth-Kühne, A.; Gottstein, S.; Geisen, U.; Schenk, J.; Scholz, U.; et al. Prognostic Factors for Remission of and Survival in Acquired Hemophilia A (AHA): Results from the GTH-AH 01/2010 Study. Blood 2015, 125, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Kessler, C.M.; Ma, A.D.; Al-Mondhiry, H.A.B.; Gut, R.Z.; Cooper, D.L. Assessment of Acquired Hemophilia Patient Demographics in the United States: The Hemostasis and Thrombosis Research Society Registry. Blood Coagul. Fibrinolysis 2016, 27, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Knoebl, P.; Marco, P.; Baudo, F.; Collins, P.; Huth-Kühne, A.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Lévesque, H. Demographic and Clinical Data in Acquired Hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). J. Thromb. Haemost. 2012, 10, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Borg, J.Y.; Guillet, B.; Le Cam-Duchez, V.; Goudemand, J.; Lévesque, H. Outcome of Acquired Haemophilia in France: The Prospective SACHA (Surveillance Des Auto AntiCorps Au Cours de l’Hémophilie Acquise) Registry. Haemophilia 2013, 19, 564–570. [Google Scholar] [CrossRef]
- Onwubiko, I.; Kasperek, G.; Laforest, R.A.; Philip, S.G.; Kuriakose, P.; Otrock, Z.K. Predictors of Response and Outcome of Patients with Acquired Haemophilia A. Haemophilia 2020, 26, e244–e246. [Google Scholar] [CrossRef]
- Jayakar, J.P.; O’Neill, N.; Yan, M.; Nisenbaum, R.; Garvey, M.B.; Teitel, J.; Sholzberg, M. Retrospective Review of Acquired Haemophilia A from the Largest Canadian Haemophilia Treatment Centre. Haemophilia 2018, 24, e383–e387. [Google Scholar] [CrossRef]
- Anaya, J.M.; Shoenfeld, Y.; Rojas-Villarraga, A.; Levy, R.A.; Cervera, R. Autoimmunity: From Bench to Bedside; El Rosario University Press: Bogotá, Colombia, 2013. [Google Scholar]
- Pisetsky, D.S. Pathogenesis of Autoimmune Disease. Nat. Rev. Nephrol. 2023, 19, 509–524. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA Variation and Disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef]
- André, S.; Meslier, Y.; Dimitrov, J.D.; Repessé, Y.; Kaveri, S.V.; Lacroix-Desmazes, S.; Dasgupta, S. A Cellular Viewpoint of Anti-FVIII Immune Response in Hemophilia A. Clin. Rev. Allergy Immunol. 2009, 37, 105–113. [Google Scholar] [CrossRef]
- Oldenburg, J.; Zeitler, H.; Pavlova, A. Genetic Markers in Acquired Haemophilia. Haemophilia 2010, 16, 41–45. [Google Scholar] [CrossRef]
- Oldenburg, J.; Picard, J.K.; Schwaab, R.; Brackmann, H.H.; Tuddenham, E.G.; Simpson, E. HLA Genotype of Patients with Severe Haemophilia A Due to Intron 22 Inversion with and without Inhibitors of Factor VIII. Thromb. Haemost. 1997, 77, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.; Delev, D.; Lacroix-Desmazes, S.; Schwaab, R.; Mende, M.; Fimmers, R.; Astermark, J.; Oldenburg, J. Impact of Polymorphisms of the Major Histocompatibility Complex Class II, Interleukin-10, Tumor Necrosis Factor-α and Cytotoxic T-Lymphocyte Antigen-4 Genes on Inhibitor Development in Severe Hemophilia A. J. Thromb. Haemost. 2009, 7, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.; Ollier, W.; Pepper, L.; Cumming, A.; Keeney, S.; Goodeve, A.; Colvin, B.; Hill, F.; Preston, F.; Peake, I. HLA Class II Profile: A Weak Determinant of Factor VIII Inhibitor Development in Severe Haemophilia A. Thromb. Haemost. 1997, 77, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Uchanska-Ziegler, B.; Loll, B.; Fabian, H.; Hee, C.S.; Saenger, W.; Ziegler, A. HLA Class I-Associated Diseases with a Suspected Autoimmune Etiology: HLA-B27 Subtypes as a Model System. Eur. J. Cell Biol. 2012, 91, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, R.; Leelayuwat, C.; Gaudieri, S.; Tay, G.; Hui, J.; Cattley, S.; Martinez, P.; Kulski, J. Genomics of the Major Histocompatibility Complex: Haplotypes, Duplication, Retroviruses and Disease. Immunol. Rev. 1999, 167, 275–304. [Google Scholar] [CrossRef]
- Hollenbach, J.A.; Oksenberg, J.R. The Immunogenetics of Multiple Sclerosis: A Comprehensive Review. J. Autoimmun. 2015, 64, 13–25. [Google Scholar] [CrossRef]
- Bettencourt, A.; Carvalho, C.; Leal, B.; Brás, S.; Lopes, D.; Martins da Silva, A.; Santos, E.; Torres, T.; Almeida, I.; Farinha, F.; et al. The Protective Role of HLA-DRB1(∗)13 in Autoimmune Diseases. J. Immunol. Res. 2015, 2015, 948723. [Google Scholar] [CrossRef]
- Wieland, I.; Wermes, C.; Eifrig, B.; Holstein, K.; Pollmann, H.; Siegmund, B.; Bidlingmaier, C.; Kurnik, K.; Nimtz-Talaska, A.; Niekrens, C.; et al. Inhibitor-Immunology-Study. Different HLA-Types Seem to Be Involved in the Inhibitor Development in Haemophilia A. Hamostaseologie 2008, 28, S26–S28. [Google Scholar]
- van der Helm-van Mil, A.H.M.; Huizinga, T.W.J.; Schreuder, G.M.T.; Breedveld, F.C.; de Vries, R.R.P.; Toes, R.E.M. An Independent Role of Protective HLA Class II Alleles in Rheumatoid Arthritis Severity and Susceptibility. Arthritis Rheum. 2005, 52, 2637–2644. [Google Scholar] [CrossRef]
- Muñiz-Castrillo, S.; Vogrig, A.; Honnorat, J. Associations between HLA and Autoimmune Neurological Diseases with Autoantibodies. Autoimmun. Highlights 2020, 11, 2. [Google Scholar] [CrossRef]
- Miyadera, H.; Ohashi, J.; Lernmark, A.; Kitamura, T.; Tokunaga, K. Cell-Surface MHC Density Profiling Reveals Instability of Autoimmunity-Associated HLA. J. Clin. Investig. 2015, 125, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Manczinger, M.; Kemény, L. Peptide Presentation by HLA-DQ Molecules Is Associated with the Development of Immune Tolerance. PeerJ 2018, 6, e5118. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, E.; Breedveld, F.C.; de Vries, R.R. HLA Association with Autoimmune Disease: A Failure to Protect? Rheumatology 2000, 39, 1060–1066. [Google Scholar] [CrossRef]
- Praditpornsilpa, K.; Kupatawintu, P.; Mongkonsritagoon, W.; Supasyndh, O.; Jootar, S.; Intarakumthornchai, T.; Pongskul, C.; Prasithsirikul, W.; Achavanuntakul, B.; Ruangkarnchanasetr, P.; et al. The Association of Anti-r-HuEpo-Associated Pure Red Cell Aplasia with HLA-DRB1*09-DQB1*0309. Nephrol. Dial. Transplant. 2009, 24, 1545–1549. [Google Scholar] [CrossRef]
- Kucuksezer, U.C.; Aktas Cetin, E.; Esen, F.; Tahrali, I.; Akdeniz, N.; Gelmez, M.Y.; Deniz, G. The Role of Natural Killer Cells in Autoimmune Diseases. Front. Immunol. 2012, 12, 622306. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liang, S.; Zhang, C. NK Cells in Autoimmune Diseases: Protective or Pathogenic? Front. Immunol. 2021, 12, 624687. [Google Scholar] [CrossRef]
- Benyamine, A.; Magalon, J.; Sabatier, F.; Lyonnet, L.; Robert, S.; Dumoulin, C.; Morange, S.; Mazodier, K.; Kaplanski, G.; Reynaud-Gaubert, M.; et al. Natural Killer Cells Exhibit a Peculiar Phenotypic Profile in Systemic Sclerosis and Are Potent Inducers of Endothelial Microparticles Release. Front. Immunol. 2018, 9, 1665. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.; Arnold, D.M.; Coller, B.; James, P.; Neunert, C.; Lillicrap, D. ISTH/SSC Bleeding Assessment Tool: A Standardized Questionnaire and a Proposal for a New Bleeding Score for Inherited Bleeding Disorders. J. Thromb. Haemost. 2010, 8, 2063–2065. [Google Scholar] [CrossRef]
- Mingot-Castellano, M.E.; Pardos-Gea, J.; Haya, S.; Bastida-Bermejo, J.M.; Tàssies, D.; Marco-Rico, A.; Núñez, R.; García-Candel, F.; Fernández-Sanchez de Mora, M.C.; Soto, I.; et al. Management of Acquired Hemophilia A: Results from the Spanish Registry. Blood Adv. 2021, 5, 3821–3829. [Google Scholar] [CrossRef]
- Closa, L.; Vidal, F.; Herrero, M.J.; Caro, J.L. Design and Validation of a Multiplex KIR and HLA Class I Genotyping Method Using Next Generation Sequencing. Front. Immunol. 2018, 9, 2991. [Google Scholar] [CrossRef]
- Closa, L.; Vidal, F.; Herrero, M.J.; Caro, J.L. Distribution of Human Killer Cell Immunoglobulin-like Receptors and Ligands among Blood Donors of Catalonia. HLA 2020, 95, 179–188. [Google Scholar] [CrossRef]
- Enrich, E.; Campos, E.; Martorell, L.; Herrero, M.J.; Vidal, F.; Querol, S.; Rudilla, F. HLA-A, -B, -C, -DRB1, and -DQB1 Allele and Haplotype Frequencies: An Analysis of Umbilical Cord Blood Units at the Barcelona Cord Blood Bank. HLA 2019, 94, 347–359. [Google Scholar] [CrossRef]
- Closa, L.; Vidal, F.; Herrero, M.J.; Caro, J.L. High-Throughput Genotyping of HLA-G, HLA-F, MICA, and MICB and Analysis of Frequency Distributions in Healthy Blood Donors from Catalonia. HLA 2021, 97, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Closa, L.; Xicoy, B.; Zamora, L.; Estrada, N.; Colomer, D.; Herrero, M.J.; Vidal, F.; Alvarez-Larrán, A.; Caro, J.L. Natural Killer Cell Receptors and Ligand Variants Modulate Response to Tyrosine Kinase Inhibitors in Patients with Chronic Myeloid Leukemia. HLA 2022, 99, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Nguyen, V.H.; Ichimura, H.; Pham, T.T.; Nguyen, C.H.; Pham, T.V.; Elbadry, M.I.; Yoshioka, K.; Tanaka, J.; Trung, L.Q.; et al. A Functional Polymorphism in the NKG2D Gene Modulates NK-Cell Cytotoxicity and Is Associated with Susceptibility to Human Papilloma Virus-Related Cancers. Sci. Rep. 2016, 6, 39231. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.P. Confidence Intervals That Match Fisher’s Exact or Blaker’s Exact Tests. Biostatistics 2010, 11, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.; Jimenez-Yuste, V.; Hernandez-Navarro, F.; Villar, A. Acquired haemophilia: Review and meta-analysis focused on therapy and prognostic factors. Br. J. Haematol. 2003, 121, 21–35. [Google Scholar] [CrossRef]
- Green, D.; Lechner, K. A survey of 215 non-hemophilic patients with inhibitors to Factor VIII. Thromb. Haemost. 1981, 30, 200–203. [Google Scholar] [CrossRef]

{kind=link}
| A. General Characteristics of the AHA Cohort | ||
| Sex, n (%) | Male | 27 (55.1) |
| Female | 22 (44.8) | |
| Male: female ratio | 1:0.81 | |
| Age at diagnosis (years) | Median (IQR) | 74 (59–79) |
| Cause of bleeding at onset, n (%) * | Spontaneous | 41 (83.7) |
| Surgery | 2 (4.1) | |
| Trauma | 3 (6.1) | |
| Peripartum | 1 (2.0) | |
| ISTH bleeding score * | Median (IQR) | 8 (6–11) |
| Males | 8.8 | |
| Females | 7.9 | |
| Remission status at data capture, n (%) | Complete | 42 (85.7) |
| Partial | 6 (12.2) | |
| No remission | 1 (2.0) | |
| Time to complete remission (days) † | Median (IQR) | 175 (84–266) |
| Death at data collection | Overall mortality, n (%) | 13 (26.5) |
| AHA specific, n (%) ‡ | 6 (12.2) | |
| B. Laboratory Parameters of the AHA Cohort at Diagnosis | ||
| Hemoglobin (g/dL) | Median (IQR) | 6.8 (6.0–8.5) |
| aPTT (s) § | Median (IQR) | 76 (62–90.8) |
| FVIII:C (%) | Median (IQR) | 1 (0.3–3.6) |
| FVIII level category, n (%) | Severe (≤1%) | 28 (57.1) |
| Moderate (>1 to ≤5%) | 12 (24.5) | |
| Mild (>6 to ≤50%) | 9 (18.4) | |
| Inhibitor titer (BU/mL) | Median (IQR) | 18 (5–62) |
| Inhibitor titer category, n (%) | 0 to ≤10 BU/mL | 19 (38.8) |
| >10 to ≤100 BU/mL | 22 (44.9) | |
| >100 to ≤1000 BU/mL | 8 (16.3) | |
| Inhibitor kinetics type, n (%) | Type I | 23 (46.9) |
| Type II | 26 (53.1) | |
| Allele | Allele Count in AHA Cohort | Allele Freq (%) in AHA Cohort | Allele Count in Reference Sample * | Allele Freq (%) in Reference Sample * | Fisher p-Value | Odds Ratio (95% CI) |
|---|---|---|---|---|---|---|
| A*03:01 | 3 | 3.06 | 22 | 11.00 | 0.024 | 0.26 (0.06–0.85) |
| DRB1*13:03 | 7 | 7.14 | 2 | 1.00 | 6.82 × 103 | 7.56 (1.64–51.40) |
| Rs1049174 Genotype | Haplotype | Count in AHA Cohort | Freq (%) in AHA Cohort | Count in IBS * | Freq (%) in IBS * | Fisher p-Value | Odds Ratio (95% CI) | Fisher p-Value for Global Association |
|---|---|---|---|---|---|---|---|---|
| CC | HNK/HNK | 0 | 0.00 | 10 | 9.35 | 0.031 | 0 (0–0.89) | 0.012 |
| CG | HNK/LNK | 29 | 59.18 | 42 | 39.25 | 0.024 | 2.23 (1.09–4.58) | |
| GG | LNK/LNK | 20 | 40.82 | 55 | 51.40 | 0.232 | 0.65 (0.32–1.34) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardos-Gea, J.; Martin-Fernandez, L.; Closa, L.; Ferrero, A.; Marzo, C.; Rubio-Rivas, M.; Mitjavila, F.; González-Porras, J.R.; Bastida, J.M.; Mateo, J.; et al. Key Genes of the Immune System and Predisposition to Acquired Hemophilia A: Evidence from a Spanish Cohort of 49 Patients Using Next-Generation Sequencing. Int. J. Mol. Sci. 2023, 24, 16372. https://doi.org/10.3390/ijms242216372
Pardos-Gea J, Martin-Fernandez L, Closa L, Ferrero A, Marzo C, Rubio-Rivas M, Mitjavila F, González-Porras JR, Bastida JM, Mateo J, et al. Key Genes of the Immune System and Predisposition to Acquired Hemophilia A: Evidence from a Spanish Cohort of 49 Patients Using Next-Generation Sequencing. International Journal of Molecular Sciences. 2023; 24(22):16372. https://doi.org/10.3390/ijms242216372
Chicago/Turabian StylePardos-Gea, Jose, Laura Martin-Fernandez, Laia Closa, Ainara Ferrero, Cristina Marzo, Manuel Rubio-Rivas, Francesca Mitjavila, José Ramón González-Porras, José María Bastida, José Mateo, and et al. 2023. "Key Genes of the Immune System and Predisposition to Acquired Hemophilia A: Evidence from a Spanish Cohort of 49 Patients Using Next-Generation Sequencing" International Journal of Molecular Sciences 24, no. 22: 16372. https://doi.org/10.3390/ijms242216372