1. Introduction
Despite extensive basic and clinical research in the last two decades, the therapy of malignant gliomas remains almost unchanged since 2005 and provides only palliation [
1,
2,
3,
4]. Therapy failure can be attributed to the histopathological features, the presence of enzymes that repair the lesions introduced by radio- and chemotherapy, detoxifying proteins such as MGMT (O6-methylguanine-DNA methyltransferase), and the presence of tumor cells with features similar to neural stem cells (SLGCs) [
5,
6,
7,
8,
9]. SLGCs were isolated from higher grade gliomas, including glioblastoma multiforme (GBM) and the gliosarcoma [
4,
6,
7,
8,
9,
10]. Standard GBM and GS therapy consists of surgical intervention followed by fractionated radiation and chemotherapy with the alkylating compound temozolomide [
1,
4,
11,
12,
13]. The efficacy of TMZ (Temozolomide) treatment is largely related to the expression of MGMT, which in turn is regulated by the methylation of the MGMT gene promoter [
1,
4,
5,
6]. Innovative strategies that try to improve the standard therapy assess the impact of the inhibitors of dysregulated oncogenic proteins and pathways or evaluate the potential benefits of immunotherapies [
14]. Whether these therapy concepts might improve GBM therapy remains unclear, since GBM resiliency is based on a complex interrelation between molecular and cellular heterogeneity and the hypoxic and metabolic characteristics of the microenvironment. In particular, the specific properties of SLGCs and their adaptive plasticity appear crucial to therapy failure [
4,
5,
6,
8,
9].
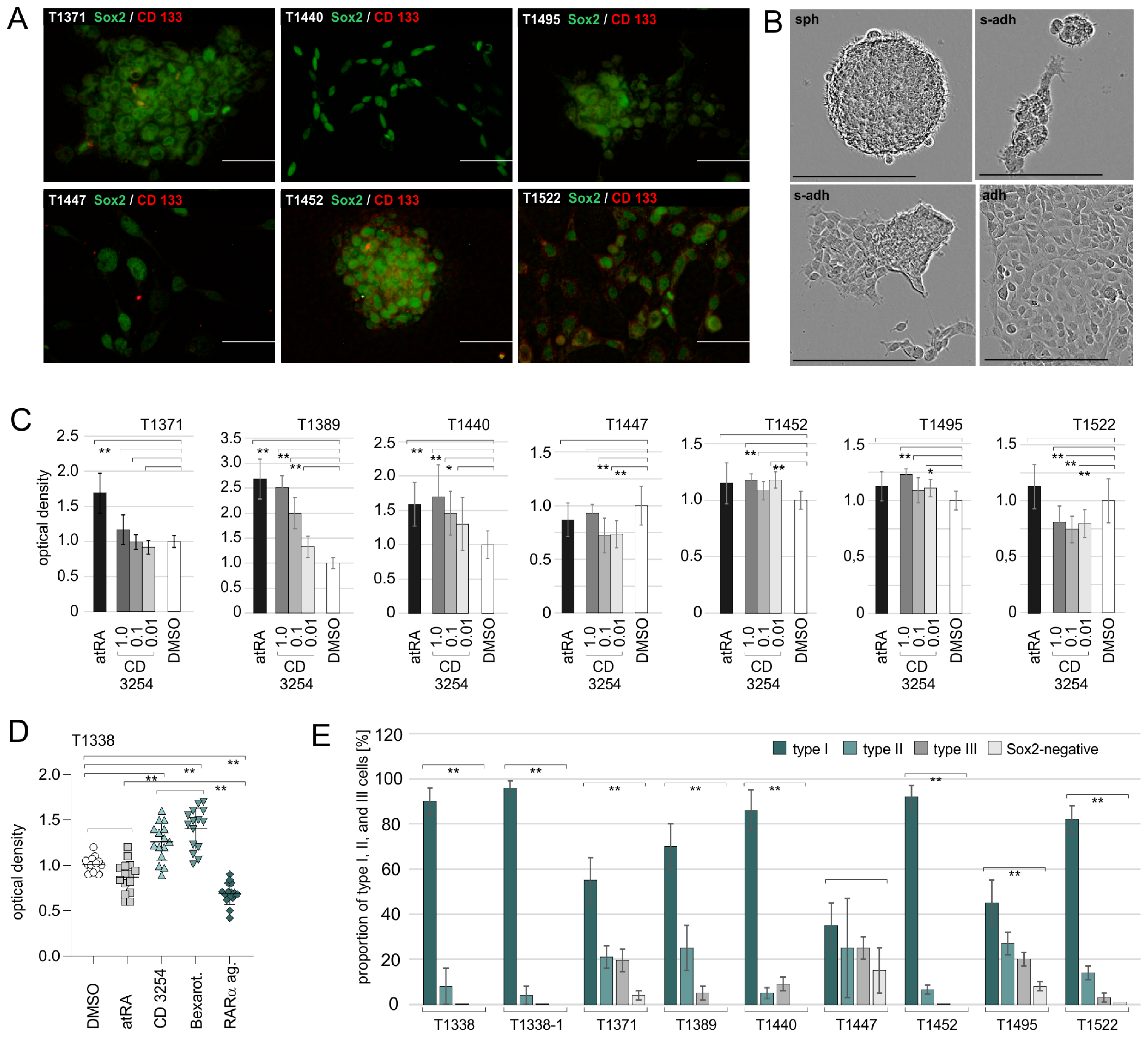
Similar to normal neural stem cells, SLGCs establish a cellular hierarchy consisting of the stem cell and more differentiated progenitors. The glioma stem cell (type I cell) expresses high levels of the pluripotency factor Sox2 (Sex Determining Region Y)-Box 2), which is crucial for the maintenance of stemness and contributes to the malignant phenotype [
9,
15,
16]. Sox2+/CD133− type I cells differentiate into Sox2+/CD133+ progenitors (type II cells), which in turn generate CD133-negative cells with reduced Sox2 expression (type III cells). Further differentiation results in the production of non-SLGCs, which account for the bulk of the tumor cells [
6,
9,
17]. Since the more differentiated GBM and GS cells are more sensitive to the standard therapy than the SLGCs and type I cells in particular [
5,
6,
17], the induction of differentiation might improve the GBM and GS therapy.
Natural components regulating the differentiation of stem cells and progenitors in the neural system include the vitamin A derivative retinoic acid (RA) [
18,
19]. The RA isomers all-trans RA (atRA) and 9cis RA (9cRA) mediate pleiotropic effects in development and homeostasis. The RA signal is transduced by the transcription factors RAR (retinoic acid receptor) and RXR (retinoid-X receptor), in which 9cRA activates both receptor types and atRA acts solely via RAR [
20]. Three distinct RAR and RXR genes are present in the human genome that code for the isotypes RAR α, β, and γ and RXR α, β, and γ, respectively [
20]. Importantly, RXR isotypes form heterodimers with multiple partners, which, in the case of the PPAR (peroxisome-proliferator-activated receptor)/RXR heterodimers, are permissive for RXR-selective ligands (rexinoids), whereas others, such as the RAR/RXR heterodimers, require the binding of an agonist to the partner of RXR [
20,
21,
22]. Evidence has been provided that the imbalance in RAR isotype expression and aberrant expression of RA signaling molecules will contribute to brain tumor development [
18,
23,
24], suggesting that the normalization of RA signaling could be beneficial. Indeed, the combination of atRA and vitamin D, or lithium, metformin, and a PPARγ agonist, might improve GBM therapy [
25,
26]. However, the biological effects of RA on GBM or GS cells are poorly understood. Specifically, whether RA could induce the differentiation of SLGCs or if the potential effects of RA would be transduced by RXR-non-permissive or RXR-permissive heterodimers remains unclear [
20].
In the present study, which is a continuation of our previous work [
7], we aimed to analyze whether the activation of RAR-dependent or RAR-independent RXR signaling would have an impact on SLGCs from human glioma specimens. First, we compared the responsiveness of six GBM- and two GS-derived SLGC lines to RAR- and RXR-selective agonists. To further decipher the contribution of RAR-dependent and RAR-independent signaling, the T1338-1 subclone, which primarily consisted of type I cells, displayed good responsiveness to retinoids and rexinoids, and lacked the expression of MGMT, was chosen for further experiments. These included CRISPR/Cas9-mediated editing of the three RAR and RXR genes as well as the subsequent characterization of the clones and their responsiveness to retinoids, rexinoids, and TMZ. We show that the functional
knockout of one or two of the RAR (or RXR) genes is possible in the respective RA-responsive SLGC line and that the degree of stemness is not affected. Moreover, we provide evidence that RAR-dependent RXR signaling can initiate differentiation, whereas RAR-independent RXR signaling rather mediates pro-proliferative and pro-survival effects, which might be associated with SLGC metabolism.
3. Discussion
Evidence has accumulated that an imbalance of retinoic acid (RA) metabolism and RAR signaling might contribute to the initiation and progression of malignant primary brain tumors [
18,
23,
24]. Moreover, the sequestration of RA in the cytoplasm of GBM cells by the cellular retinoic acid-binding protein 2 (CRABP2) appeared to be related to the poor survival of GBM patients [
42]. A very recent study comparing data from The Cancer Genome Atlas (TCGA) indicated that pathways associated with RA metabolism may play an important role in glioma etiology, suggesting that the future exploration of these genes and pathways may influence the development of novel therapies for the incurable disease [
43]. Indeed, RA exerts neuroprotective functions, ensures the physiological structural integrity of the neurovascular unit in the brain, and may eradicate cancer stem cells in certain human cancers, including renal, lung, colorectal, and ovarian cancer [
44,
45,
46]. Whether RA would deplete cancer stem cells from primary human brain tumors is less clear. In our previous work [
7], we have shown that only a subpopulation of cancer stem cells from human GBM and GS specimens (referred to as SLGCs in the present manuscript) enter differentiation in response to RA and that the RA signal is transduced via RARα and RARγ in these cases [
7]. Our previous data also suggested a contribution of RAR-independent RXR signaling to the treatment outcome, which we aimed to decipher in the present work.
IDH1-positive SLGC lines from six GBM and two GS specimens were investigated. Confirming our previous results, the effects of atRA varied between the SLGC lines. SLGCs from two GBMs and one GS showed increased proliferation rates after treatment with 1 µM atRA, and one GBM-derived (T1338) and one GS-derived (T1447) SLGC line displayed growth inhibition. The results for distinct biological and technical replicates, in particular T1338, T1440, and T1452, revealed substantial variations, which appeared related to the variations in growth behavior. Another complication is the fact that atRA may undergo isomerization into 9cRA in the medium [
38], suggesting that the effects of atRA might have been mediated by RAR/RXR heterodimers or distinct RXR heterodimers, which are permissive to 9cRA signaling [
21,
22]. The RXR-selective agonist CD3254, which does not affect the activity of RAR isotypes [
7], exerted a significant pro-proliferative effect in most SLGC lines and inhibited proliferation in two cases. Moreover, CD3254 did not significantly potentiate the growth inhibitory effects of the RAR pan-agonist CD0270 in most SLGC lines, suggesting that the pro-proliferative or pro-survival effects are exerted by RAR-independent pathways.
We isolated T1338 subclones that are homozygous for the p53 wildtype from the original heterogeneous T1338 culture and tested their growth behavior and atRA responsiveness. The growth behavior of these subclones differed, and most of them displayed a semi-adherent (s-adh) phenotype, i.e., they primarily formed adherent aggregates. When treated with 1 µM CD3254 (or bexarotene), the incorporation of BrdU was significantly augmented by both rexinoids, suggesting that rexinoids exert a pro-proliferative and/or pro-survival effect. Altogether, the RAR pan-agonist but not atRA mediated a significant reduction in the proliferation of the T1338 subclones. Substantial variations were observed between the biological and technical replicates. These are related to variations in growth behavior, which cannot be fully controlled, and the properties of the retinoids [
7,
38]. Major complications are that single cell suspensions of T1338 cells form adherent or floating aggregates within two days, that these aggregates fuse, and that single cells shuttle between the aggregates. Another complication is that the cells in the centers of aggregates will not receive the same doses of retinoids as the cells on the periphery. Treatment of distinct T1338 p53
WT/WT clones with RAR isotype-selective retinoids revealed that the atRA signal is transduced through RARα and RARγ. This is in keeping with the observation that the RARβ promoter is hypermethylated and associated with the repressive chromatin mark H3K9me2/3. In this context, it should be mentioned that histone lysine methyltransferase SETDB1, which mediates di-/tri-methylation of the histone H3 in the lysine residue K9, is thought to function as a common denominator of gene regulation in several cancer types, in which most data point to a pro-oncogenic role of SETDB1 [
31].
The significance of the conclusions on RAR-dependent and RAR-independent RXR-signaling in SLGCs described above is limited by several parameters: first, several SLGCs form adherent and floating aggregates, which hampers the reproducibility of the experiments and the potency of retinoid treatment. Second, the use of the antibodies directed against the various RAR and RXR isotypes is limited by their K
Ds and specificities. Third, the RAR isotype-selective agonists are no longer selective when used at higher concentrations [
7], and RXR isotype-selective ligands are not yet available. Fourth, though the SLGC lines are grown in a serum-free medium, it cannot be fully excluded that the medium component BIT (serum albumin-insulin-transferrin) contains traces of retinoids. Therefore, we decided to apply CRISPR/Cas9-mediated editing [
33]. We chose a lentiviral vector [
34] to overcome the low transfection efficacies associated with the SLGC phenotype. Despite the problems illustrated above, we selected the T1338 p53
WT/WT subclone T1338-1 for the editing approaches because of its high degree of stemness, the very high content of type I cells, and the good responsiveness to RAR- and RXR-selective ligands and TMZ.
The editing approaches were designed such that the sgRNAs targeted the exons of the RARA, RARB, RARG, RXRA, RXRB, and RXRG genes, which encode the first of the two zinc fingers. This strategy was chosen since the first zinc finger encompasses the so-called P-box, which mediates the recognition and binding of RARs and RXRs to their cognate response elements [
20]. We used a lentiviral vector, which allowed for puromycin selection after the infection of the cells with the recombinant viruses. The efficacy of the puromycin selection, however, was hampered by the growth behavior of the T1338-1 cells. As could have been expected, a substantial number of wildtype clones were isolated from limited dilution experiments in addition to the edited clones. The editing of RARA and RXRA was highly efficient, as several isolated clones carried biallelic functional knockouts of the RARA and RXRG genes, respectively. The editing efficiency was considerably reduced for RARB and RARG, but also RXRB and RXRG. Moreover, most clones obtained from the RARB editing approach harbored RARB wildtype sequences. Since the wildtype sequences could not be removed by several additional rounds of limited dilution assays, this indicated that RARB editing occurred only in one allele in most of these clones. Considering the hypermethylation of the RARβ promoter and the increased proportion of the repressive mark H3K9me2/3, it could be speculated that the RARB gene was less accessible because of a comparatively closed chromatin state. Indeed, the efficacy of Cas9-mediated gene editing is dependent on chromatin accessibility [
47]. Unexpectedly, the targeting of the RARA gene resulted in >90% of the cases having a homozygous insertion of a single nucleotide, which was an adenosine-monophosphate in 11 out of 12 cases and a thymidine-monophosphate in one single case. The editing approach of the RXRA gene was similarly efficient, with a slight preference for a homozygous insertion of a thymidine-monophosphate; but deletions of up to three nucleotides were also frequently observed. Biallelic editing of the RARG, RXRB, and RXRG genes was possible, but with a lower efficiency. The simultaneous editing of two RAR isotypes was possible, and signs of growth reduction did not become evident in the RAR double knockouts. In the case of the RXR editing approaches, only one clone (RXR H6αγ) with a full knockout of the RXRA and RXRG genes was obtained, and one other clone (RXR C5αγ) most likely harbored a biallelic knockout of the RXRA gene plus a monoallelic knockout of RXRG. In contrast to the clones with a functional knockout of only one RXR isotype, the clone RXR H6αγ displayed impaired growth. This was not observed for RXR C5αγ, which still carried one functional RXRG gene. Both clones, particularly RXR H6αγ, showed reduced expression of the LDHA. Considering that the RXR isotypes form heterodimers with several nuclear receptors, including the PPARs, which function as metabolic sensors and regulate carbohydrate and/or lipid metabolism [
21,
22], the reduced expression of LDHA could indicate that RXR isotypes are necessary for the metabolic adaptation of SLGCs. Notably, the expression of PMK2 was high in T1338 cells and remained unaffected by RXR editing.
The T1338 clones with functional inactivation of one RAR isotype or one RXR isotype as well as the clones RXR H6αγ and RXR C5αγ were selected for further experiments. The first central aim was the analysis of stemness. This was approached by double stains using antibodies against Sox2 and CD133. Except for one RAR (RAR F2γ) and two RXR clones (RXR C10α, RXR F4α), the cultures of the various RAR and RXR clones consisted primarily of Sox2+ cells that did not express CD133. Since the Sox2 signal was very strong in >95% of the cells, the respective cultures contained almost solely type I cells. Similar to the T1338 mother culture, all clones expressed the neuronal protein Tau and low levels of the glial fibrillary protein GFAP. In the cases of the RXR knockout clones, however, the expression levels of Tau and GFAP were increased, even in the presence of high levels of Sox2. This indicated that a high degree of stemness does not prevent the expression of Tau and GFAP and that the inactivation of one RXR isotype might result in higher levels of these neural proteins. Treatment with 1µM atRA/1 mM cAMP further increased the Tau and GFAP levels in the respective RXR clones and drastically decreased cell numbers. In the cases of the RAR clones, in which the effects of 1 µM atRA were assessed in the absence of cAMP, the treatment with 1 µM atRA reduced the cell numbers and moderately increased the expression of Tau and GFAP.
Experiments comparing the proliferation and growth of clones with edited RAR or RXR genes revealed substantial heterogeneity between clones with the same type of editing. Significant differences were also observed between the RAR and RXR mock clones, respectively. The direct comparison of the proliferation of two RXR (or RAR) mock clones with several edited RXR (or RAR) clones suggested that the homozygous inactivation of only one RXR (or RAR) isotype does not impair the ability of T1338 cells to stably proliferate in N-medium. Solely, the full inactivation of both, RXRα and RXRγ, which was achieved in the clone RXR H6αγ, impaired growth, probably reflecting the reduced availability of RXR isotypes for the formation of RXR permissive or non-permissive heterodimers [
20,
21,
22]. In the presence of the RXR pan-agonist CD3254, a significant increase in proliferation was observed for both RXR mock clones. This pro-proliferative effect was reduced for the RXRA knockouts and absent or almost absent for the RXRB, RARG, and/or RXRA/RXRG knockouts. Hence, the knockout of one RXR isotype might be compensated by the two others, yet the limited numbers of RXR receptors might derogate the activity of several RXR-dependent nuclear receptor signaling pathways [
20,
21,
22]. This could especially alleviate the capacity of T1338 (and presumably other SLGCs) to adapt to metabolic requirements [
39,
40]. The plausibility of such a scenario is revealed by reduced LDHA levels in the double knockout RXR H6αγ. In this context, it might also be interesting that RXRβ and RXRγ null mice are viable without abnormalities typically associated with vitamin A deficiency (VAD) or vitamin A excess, whereas the RXRα knockout resulted in severe fetal VAD syndrome and death (overview in [
20]).
Several recent reports suggest a central role for RAR and RXR signaling during the specification of neural fates, neuronal differentiation, neurodegenerative disease, and brain cancer [
19,
20,
21,
22,
48,
49,
50]. In order to evaluate whether RXR signaling might improve the cytotoxic effects of TMZ, we treated the RXR mock clones with the rexinoid bexarotene. We chose bexarotene for these assays since it has been approved by the FDA for clinical applications, it possesses more favorable K
Ds than CD3254, and the effects of bexarotene derivatives have been examined in the context of GBM cell lines [
51]. Bexareotene did not deplete Sox2+ cells from the cultures of edited RXR clones or the mock controls, and 1 µM bexarotene mediated a dose-dependent pro-proliferative effect in a subset of clones. When co-applied with 25 µM TMZ, which exerted a strong cytotoxic effect, 1 µM bexarotene weakened the TMZ-mediated effect in a subset of clones. This effect was most pronounced in the clones with the RXRα/γ double knockouts and appeared otherwise independent of the type of RXR knockout. The phosphorylation of the histone variant H2AX (which generates γH2AX), which is used as a potential measure for the induction of DNA lesions [
52,
53], was detected in almost all cells that survived the five-day treatment with 25 µM TMZ. In contrast, γH2AX+ cells were nearly absent in the DMSO controls and the bexarotene-treated cultures of the edited RXR clones and the RXR mock controls. On average, the few cells that survived the five-day treatment with TMZ in the presence of bexarotene exhibited a similarly strong γH2AX signal as the survivors of the sole TMZ treatment. Finally, TMZ and bexarotene/TMZ mediated a similar strong reduction in Sox2-positive cells. Taken together, this suggests that the bexarotene signal may be transduced through any of the RXR isotypes. Moreover, bexarotene is unlikely to inhibit the growth of SLGCs or sensitize SLGCs for TMZ treatment. This is contradictory to two reports, which assessed the responsiveness of the rat glioma cell line C6 [
54] or primary GBM cells [
55] to bexarotene and postulated anti-proliferative effects. Since distinct driver mutations are present in SLGCs from distinct GBM (and GS) patients and SLGCS may exhibit adaptive plasticity [
3,
9], it cannot be excluded that bexarotene might sensitize a certain subgroup of SLGCs with specific features in their genomes and epigenomes.
4. Materials and Methods
Stem-like glioma cells (SLGCs), cell culture, and growth behavior: Experiments with cell lines derived from human tumor specimens were performed according to the Helsinki guidelines, in compliance with the national regulations for the experimental use of human material (vote 08-070 of the Ethics Commission at the University of Lübeck). SLGC (stem-like glioma cell) lines were established from human glioblastoma multiforme (GBM) or gliosarcoma (GS) [
7,
10,
27]. Tumor specimens were obtained from patients who underwent routine tumor resection and expressed their agreement to participate in the study. The classification of tumor type and grade was according to the WHO criteria and carried out by a neuropathologist. Tumor specimens were made anonymous. The cell lines and subclones derived thereof were designated “T”, followed by the code number of the tumor plus a suffix in the case of clones. SLGCs were grown in serum-free N-medium, which contained DMEM/Ham’s F12 (Biochrom; Berlin, Germany), 20% BIT 9500 serum-free supplement (PELOBiotech GmbH, Planegg, Germany), 2% of a 200 mM glutamine solution, 1% amphotericin, 1% standard penicillin/streptomycin mix, and 20 ng/mL of recombinant human EGF and bFGF (Promo Cell, Heidelberg, Germany)). Passaging was performed with trypsin/EDTA (Invitrogen, Karlsruhe, Germany). All cell cultures were incubated at 37 °C in a water-saturated atmosphere in the presence of an air–carbon dioxide (5% CO
2) mixture. Growth curves: Cells were seeded at a concentration of 1 × 10
4 cells per well in 24-well plates (Greiner, Frickenhausen, Germany). These assays were performed in pure N-medium in the absence of fibronectin, to allow for spherical growth. Counting was performed on the days indicated in the figures; harvesting and dissociation of spheres were carried out using trypsin/EDTA, Trypan Blue (Sigma-Aldrich, Munich, Germany), and Neubauer counting chambers (VWR, Haasrode, Belgium). Assays were performed in triplicate and repeated with another biological replicate, which was regrown from a cryo-stock. Growth phenotype: Monitoring of the growth of spheroids, adherent aggregates, or monolayers was performed using the Incucyte
® SX (Sartorius, Göttingen, Germany). Cells were plated at 1 × 10
4 cells per cm
2 in N-medium and observed over 12 days.
Immunocytochemistry (ICC): Cells were analyzed on glass-coverslips or 8-well chamber slides. Adherence was increased by coating the growth substrates with fibronectin (1:100 dilution in 1× PBS; Promo Cell, Heidelberg, Germany). In order to prevent cell loss, all T1338 cells (subclones mother culture and edited clones) had to be analyzed in the presence of 2% fetal calf serum (FCS), which was added to the N-medium. N-medium containing 2% FCS will not induce differentiation [
10]. Fixation was performed for 7 min at −20 °C using a mixture of ethanol–acetic acid (95:5
v:
v). Primary and secondary antibodies are listed in the
File S2 Section S6. DAPI nuclear counterstain was performed with Hoechst 33258 (Roche, Mannheim, Germany). Fluoromount-G (Southern Biotechnologies, Birmingham, USA) was used for mounting. Results were observed by fluorescence microscopy using the BZ8000 and the software BZ9000 (Keyence, Neu-Isenburg, Germany). Microphotographs of aggregates and spheres were taken as Z-stacks (pitch 0.2 to 0.5). A minimum of three independent biological replicates, including two technical replicates each, were analyzed. The quantification of the signal intensities was performed using the software QuPath (version v0.2.3). Based on the model of cellular hierarchy [
17], CD133-negative cells with high Sox2 expression are classified as type I cells; type II cells co-express Sox2 and CD133; CD133-negative cells with lower Sox2 expression are classified as type III cells. DAPI nuclear counterstain was omitted from the images prior to quantification.
Western blot analysis: Cells were harvested in 1 mL of TEN [10 mM Tris-HCl (pH 7.5), 1 mM EDTA (pH 8.0), and 150 mM NaCl]. Protein extraction was performed with a buffer containing 50 mM Tris-HCl (pH7.5), 150 mM NaCl, 10% glycerol (
v:
v), 0.5% Triton-X100 (
v:
v), 0.1% of a saturated PMSF solution (Phenylmethylsulfonylfluorid; Sigma-Aldrich, Munich, Germany), and a protease inhibitor cocktail (PIC; Roche, Mannheim, Germany). Cell debris was removed by a high-speed clearing spin. In total, 15 µg (p21
CIP1, Sox2, PKM2, LDHA), and 20 µg (MGMT, RAR and RXR antibodies) of whole cell extracts (WCE) were resolved by SDS-PAGE (Minigel Protean III, BioRad, Munich, Germany) and transferred onto nitrocellulose membranes (0.45 µm, BioRad) using the Semidry Transfer System TransBlot SD (BioRad). Electrophoresis was carried out with run buffer (25 mM Trizma Base, 192 mM glycine, 0.1% SDS); a buffer containing 48 mM Trizma Base (pH 8.3), 39 mM glycine, 0.037% SDS (
w:
v), and 20% methanol (
v:
v) was used for protein transfer. Separation of WCEs was performed on 15% resolving gels for MGMT and p21
CIP; in all other cases, resolving gels contained 10% polyacrylamide and stacking gels of 4%. The loading controls used to normalize protein expression were pan-Actin and GAPDH (glyceraldehyde 3-phosphate dehydrogenase). Primary antibodies (see
File S2 Section S6) were revealed using horseradish peroxidase- (POD-) conjugated secondary antibodies (see,
File S2 Section S6) and Super Signal
® West Dura Extended Duration Substrate (#34076; Thermo Fisher Scientific; Loughborough, UK) or Clarity Max Western ECL Substrate (#1705062, BioRad). Chemiluminescence was detected with the ChemiDoc XRS system (BioRad), and quantification was performed with the software Quantity One versions 4.6.2-4.6.6 (BioRad). Western blots included a minimum of three biological replicates, two of which were regrown from cryostocks. Depending on how much WCE was available from the individual experiments, 2–4 technical replicates were produced with the same WCE.
Methylation-specific PCR (MSP): Genomic DNA was extracted using the DNeasy Mini-Kit (#69504, Qiagen, Hilden, Germany) followed by quantitative and qualitative characterization using standard protocols. Bisulfite modification of 1 µg of DNA and subsequent purification on glass milk were performed with the CpGenome
TM DNA Modification Kit reagents (#S7820; Merck Millipore, Darmstadt, Germany) according to the manufacturer’s instructions. PCR was performed using standard protocols [
56] (for primers, see
File S2 Section S7). Nucleotides (dATP, dCTP, dGTP, dTTP, 10 mM each) were purchased from Roche (Mannheim, Germany, #04638956001), and Taq polymerase (5 units/µL plus 10× buffer) from VWR International (Haasrode, Belgium; #VWR-733-1300). PCR products were mixed with 6× orange loading dye (Thermo Fisher Scientific, Schwerte, Germany), run on 2 % TBE agarose, and quantified using the ChemiDoc XRS and the software
Quantity One (BioRad, Munich, Germany). The MSP of treated cells included a minimum of three independent technical replicates. The MGMT status of the SLGC lines was determined in >5 biological replicates.
Genomic editing using the CRISPR-Cas9 system [
33]: For single guide (sg) RNA design and exclusion of OFF targets, we applied several online tools (
http://www.rgenome.net/), including CRISPR RGEN, Cas-OFFinder, Cas-Designer, BE-Analyzer—RGEN, and the ENSEMBL Genome Browser (
https://www.ensembl.org/index.html). Cloning of the DNA coding for the sgRNA was performed using standard protocols [
56,
57]. In brief, the vector lenti-CRISPR v2 (Plasmid #52961; Addgene Cambridge, MA, USA [
34]) was cleaved with the enzyme BsmBI (#R0580S, New England Biolabs, Ipswich, MA, USA), dephosphorylated using calf intestinal alkaline phosphatase (CIAP, #18009019, Thermo Fisher Scientic), and purified by phenol/chloroform extraction and ethanol precipitation. The oligonucleotides for the sgDNA cloning were phosphorylated using den T4 PNK (#MO201S; New England Biolabs), followed by the inactivation of the enzyme. Afterward, the oligonucleotides were annealed at a concentration of 10 µM each. The annealing product was inserted into the lenti-CRISPR v2 vector using the T4 Quick ligase (#M2200S, New England Biolabs), followed by transfection into
E. coli K12 Stbl3 (#C737303, Thermo Fisher Scientific). Plasmid DNA was extracted by applying a standard alkaline lysis protocol, including phenol/chloroform extraction and ethanol precipitation [
56,
57]; correct insertion of the sgDNA was determined by means of Sanger sequencing using the BrilliantDye™ Terminator (v3.1) Cycle Sequencing Kit (100 rxn, #BRD3-100, NimaGen, Nijmegen, The Netherlands). After purification on NucleoSEQ
® columns (#740523.250, Macherey-Nagel, Düren, Germany) and denaturation in the presence of HiDi
TM Formamide (Thermo Fisher Scientic), the samples were analyzed on the Applied Biosystems 3130 Genetic Analyzer using the Sequencing Analysis Software v5.2 (Applied Biosystems, Foster City, USA). Sequence analyses were carried out by means of Chromas (Technelysium, South Brisbane, AUS) and the ENSEMBL Genome Browser (
https://www.ensembl.org/index.html). The recombinant lenti-CRISPR v2 vectors were co-transfected with two packaging plasmids (psPAX2 (Addgene plasmid #12260) and pCMV-VSV-G (Addgene plasmid #8454)) into HEK293T cells (American Type Culture, Manassas, VA, USA) using the calcium-phosphate technique. Virus preparations were purified through 0.45 µm PES Millex
® filters (Merck Millipore). Infection of the target cell line T1338-1 (referred to as T1338 MC afterward) was in the presence of Polypren (8 mg/mL, Sigma-Aldrich, 1:1,000 dilution). Selection with Puromycin (Life Technologies
TM, Carlsbad, CA, USA) was carried out at a concentration of 1 µg/mL for seven days. Limited dilution assays were used to isolate clones, which were subsequently expanded to obtain enough material for cryo-conservation and gDNA extraction (DNeasy, #69504, Qiagen, Hilden, Germany). Genomic editing was revealed by PCR-mediated amplification of the targeted exon using the respective RAR or RXR isotype-selective primer pairs, followed by purification on TBE-agarose gels and spun column chromatography (NucleoSpin Gel and PCR Clean-Up Kit, #740609.50, Macherey-Nagel). Subsequent Sanger sequencing was performed as described above, using purified PCR products or recombinant pCR
TM2.1-TOPO
® DNA; amino acid sequences were deduced using the Expasy tool that was developed by the SIB (Swiss Institute of Bioinformatics, Lausanne, Switzerland). Recombinant plasmids were generated by TA cloning (Eukaryotic TA Cloning Kit, #2474228, Invitrogen) of the purified PCR fragments into the vector pCR
TM2.1-TOPO
® (Invitrogen). For amplification of recombinant pCR
TM2.1-TOPO
®, we used E.coli K12 DH5α (initially obtained from Invitrogen); plasmid extraction was done by alkaline lysis [
56,
57]; for sequencing of pCR
TM2.1-TOPO
®, we used the primers M13-62-ex_F and M13-62-ex_R, respectively. In those cases in which the sequencing data suggested a heterogeneous cell population and/or the presence of residual wildtype sequences, additional rounds of limited dilution assays were carried out. This was necessary for the isolation of clones with edited RARβ, RARγ, RXRβ, and RXRγ genes. The editing of the RARβ gene was particularly ineffective. In spite of three additional rounds of limited dilution assays, three RARβ clones still contained RARβ wildtype sequences, indicating that biallelic editing did not occur. The primers used for editing, cloning, or sequencing are listed in the
File S2 Section S7.
Synthetic retinoids and additional reagents: Temozolomide (TMZ, Temodal
®; Essex Pharma GmbH, Munich, Germany); all-trans retinoic acid (atRA, Sigma-Aldrich, Munich, Germany); pan RAR agonist: CD0270 (Galderma Laboratories, Düsseldorf, Germany); RXR pan agonists: CD3234 (K
D 50 nM for RXRα, RXRβ, and RXRγ; Tocris Bioscience), Bexarotene (K
D 24 nM RXRβ, 25 nM RXRγ, 33 nM RXRα; Tocris Bioscience); RARα agonists: CD1556 (Galderma Laboratories); Am580 (EC50 0.3 nM RAR, 8.6 nM RARβ, 13 nM RARγ; Tocris Bioscience, Minneapolis, MN, USA); RARβ agonist: CD1886 (Galderma Laboratories); RARγ agonist: CD2640 (Galderma Laboratories). The K
Ds of the synthetic retinoids CD0270, CD1556, CD1886, and CD2640 have already been described in our previous study [
7]. All compounds were dissolved in DMSO. Notably, all assays without TMZ and/or ligands contained a final concentration of 1% DMSO.
BrdU-ELISA: These assays were performed on fibronectin-coated 96-well plates. Each experiment included a minimum of two biological replicates. Two identical plates, each encompassing eight independent values, were analyzed. The responsiveness of distinct SLGC lines was monitored on days d3, d5, and d7. For the T1338 line, which was selected for further experiments, more than 15 biological replicates (subclones; T1338-1 is referred to as MC, mother culture) were analyzed. For the clones with RAR or RXR edits, a minimum of three biological replicates were analyzed. In order to prevent cell loss, all assays with T1338 cells had to be performed in the presence of 2% fetal calf serum (FCS), which does not impact the degree of stemness in the presence of the growth factors EGF and bFGF [
10]. Coating with fibronectin (5 µg/mL; Promo Cell, Heidelberg, Germany) was at a dilution of 1:100. Analyses were initially performed using the kits marketed by Roche (Mannheim, Germany; #114446141001 or #11647229001) and continued using the BrdU Cell Proliferation Assay Kit marketed by Cell Signaling (Danvers, MA, USA, #6813S). Due to the suspension of deliveries, another BrdU kit (BrdU Cell Proliferation Assay Kit, #6813, Cell Signaling) had to be implemented into the study at later stages. All experiments were carried out according to the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP): Cells were analyzed by ChIP during their exponential growth phase. Floating spheres were harvested by centrifugation (10 min, 250× g, 4 °C), and semi-adherent and adherent cells were detached with a rubber policeman. We used the simpleChIPTM enzymatic chromatin IP Kit (Cell Signaling, Danvers, MA, USA), applying the manufacturer’s protocol with several adaptations. These concerned the treatment with the micrococcal nuclease (duration, concentration of enzyme), the sonification steps (pulses and number of sonification cycles), and the immunoprecipitation steps, which had to be optimized for each individual cell line and antibody. Reversal of the cross-link was achieved with proteinase K followed by spin column purification (kit components). Only chromatin preparations with mono- to penta-nucleosomes were used. Immunoprecipitation was preceded by a preclearing step (2–4 h on a rotator at 4 °C) using a mixture of Protein A and Protein G agarose beads (Cell Signaling). The chromatin was incubated for 4 h with the antibodies before capture was carried out with Protein G agarose beads (4 °C, overnight, on a rotator). The washing steps and release of the immunoprecipitated DNA were performed according to the simpleChIPTM enzymatic chromatin IP Kit protocol. All assays included precipitations with a suitable, unrelated IgG antibody. At least three independent PCR reactions were carried out. The number of amplification cycles was 28–32 for rpl30 and 30–35 for rarβ. PCR products were resolved on 1.6 % TBE-agarose gels and quantified using the ChemiDoc XRS and the Quantity One” software (BioRad). The primer pair rpl30 F/R was obtained from Cell Signaling (simpleChIPTM Kit); all other primers used for ChIP are listed in the File S2 Section S7. The antibodies used were αIgG (#2729, Cell Signaling), αH3 (#2650, Cell Signaling), αH3K9me2/3 (#5327, Cell Signaling), and αH3K9ac (#9674, Cell Signaling).
qRT-PCR analysis: Extraction of total RNA was performed using the RNeasy Mini-Kit (Qiagen) followed by a DNase I-treatment (Ambion, Darmstadt, Germany), and extraction with phenol-chloroform (Roth) and ethanol (Merck) precipitation was according to standard protocols [
56]. cDNA synthesis was performed with the iScript
TM Reverse Transcription Supermix for RT-qPCR kit (BioRad, Munich, Germany) followed by second strand synthesis with the qPCR Core kit for SYBR
® Green I kit (Eurogentec, Cologne, Germany) and monitoring of product synthesis with the CFX96 Real-time PCR Detection System (BioRad). Quantification of the PCR products was relative to the reference genes
gapdh,
ubiquitin ligase, and 18s
r-rna. Relative expression was calculated according to the ΔΔ-CT method. Analyses were performed in triplicate, using RNA from two biological replicates. DNA-oligonucleotides were purchased from Eurofins genomics (Ebersberg, Germany). The respective sequences have been published elsewhere [
7].
Statistical analysis was performed using GraphPad Prism version 9 (Boston, MA, USA). In order to determine significant differences in BrdU ELISA, growth curves, Western blots, immunocytochemistry analyses, and qRT-PCR analyses, a one-sided ANOVA followed by post hoc Bonferroni correction for multiple testing was used; adjusted p-values are indicated in the figures and legends to the figures. Since the KDs of the antibodies used for ChIP are not known and since the efficacy of bisulfate modification may vary, descriptive statistics were used for ChIP and MSP; mean values and standard deviations are indicated in the respective figures.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}