
Pan-Inhibition of Protein Disulfide Isomerase Caused Cell Death through Disrupting Cellular Proteostasis in Pancreatic Ductal Adenocarcinoma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
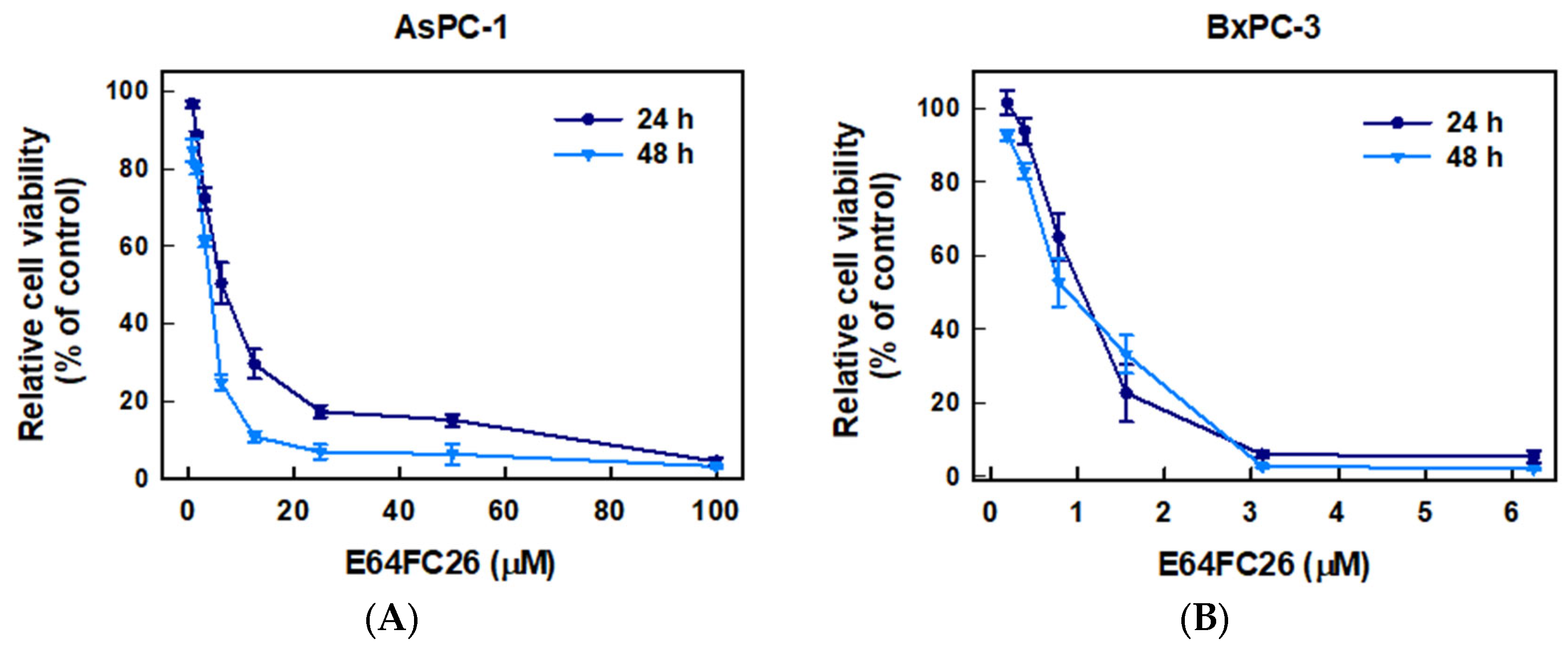
2.1. The PDI Inhibitor, E64FC26, Inhibited Proliferation of PDAC Cells
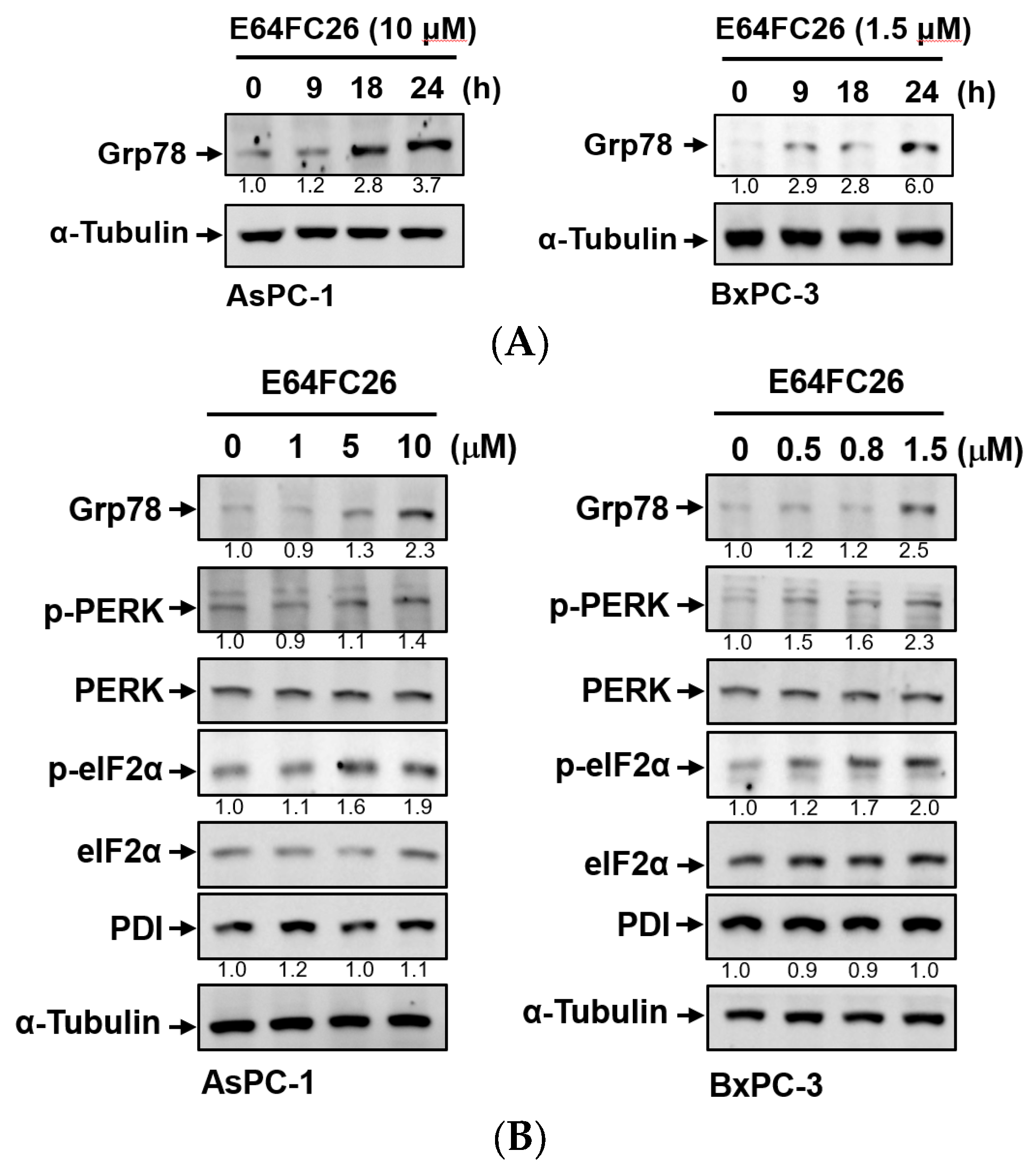
2.2. The PDI Inhibitor, E64FC26, Induced ER Stress and the UPR
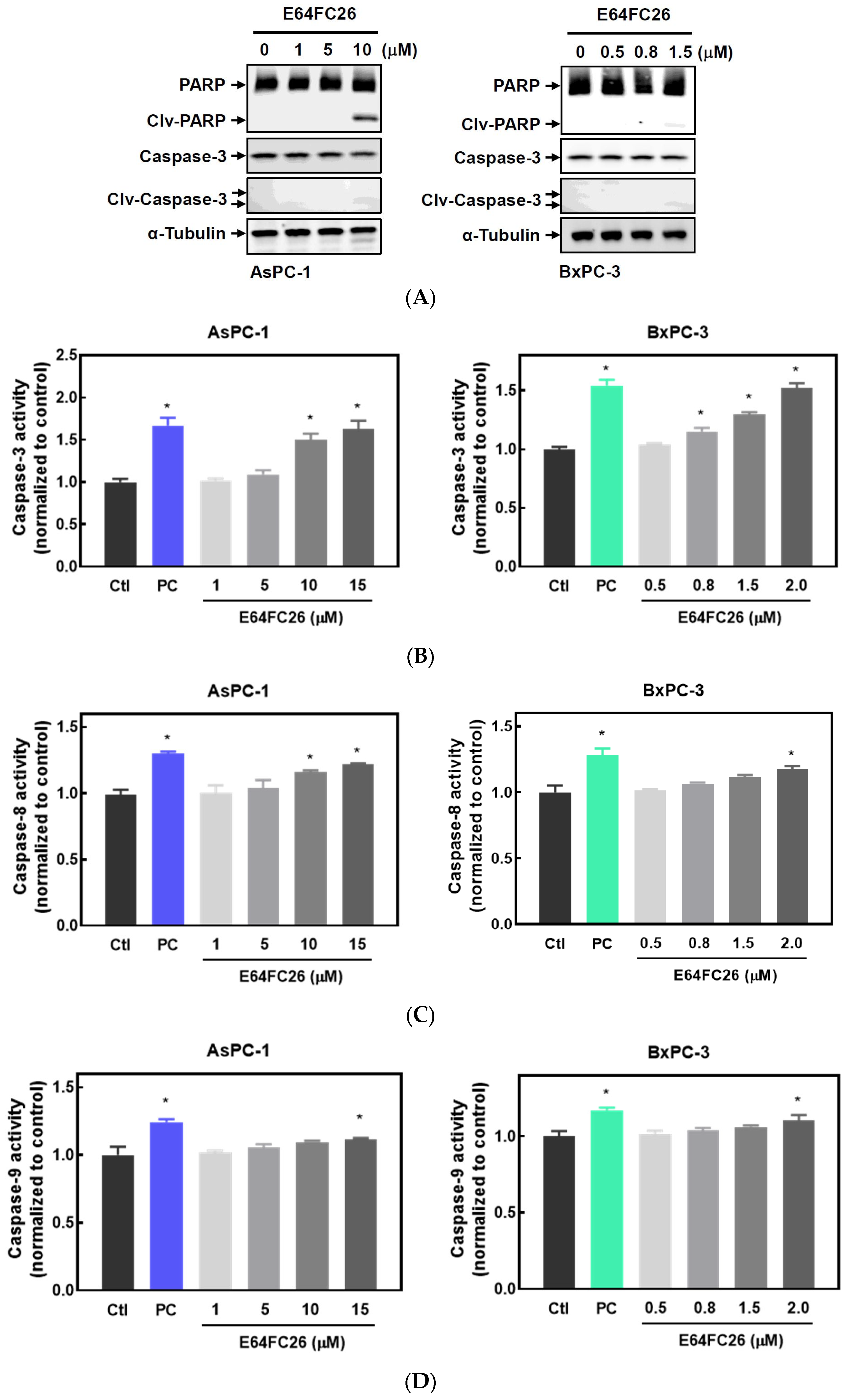
2.3. The PDI Inhibitor, E64FC26, Caused Cell Death Partially via Apoptosis
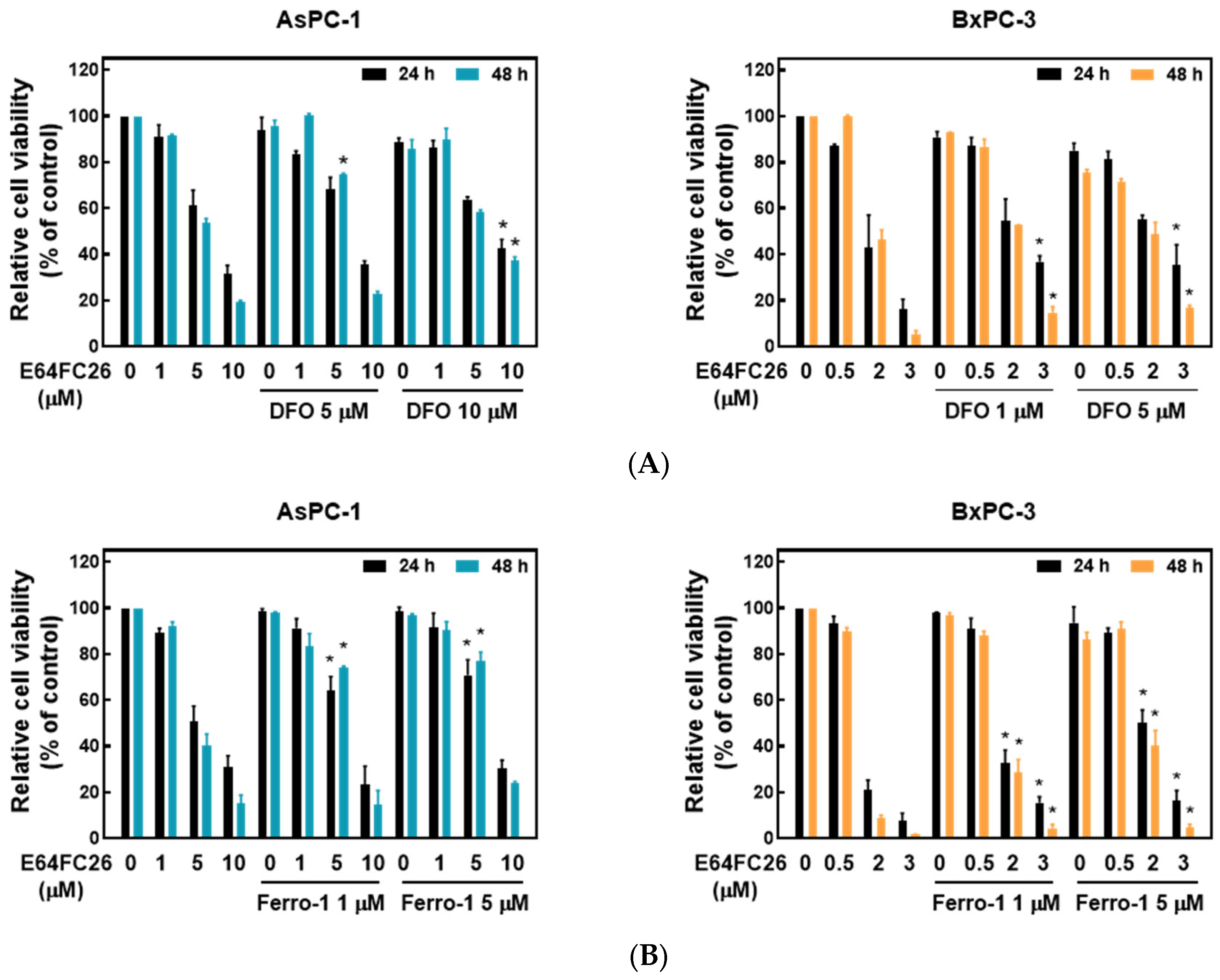
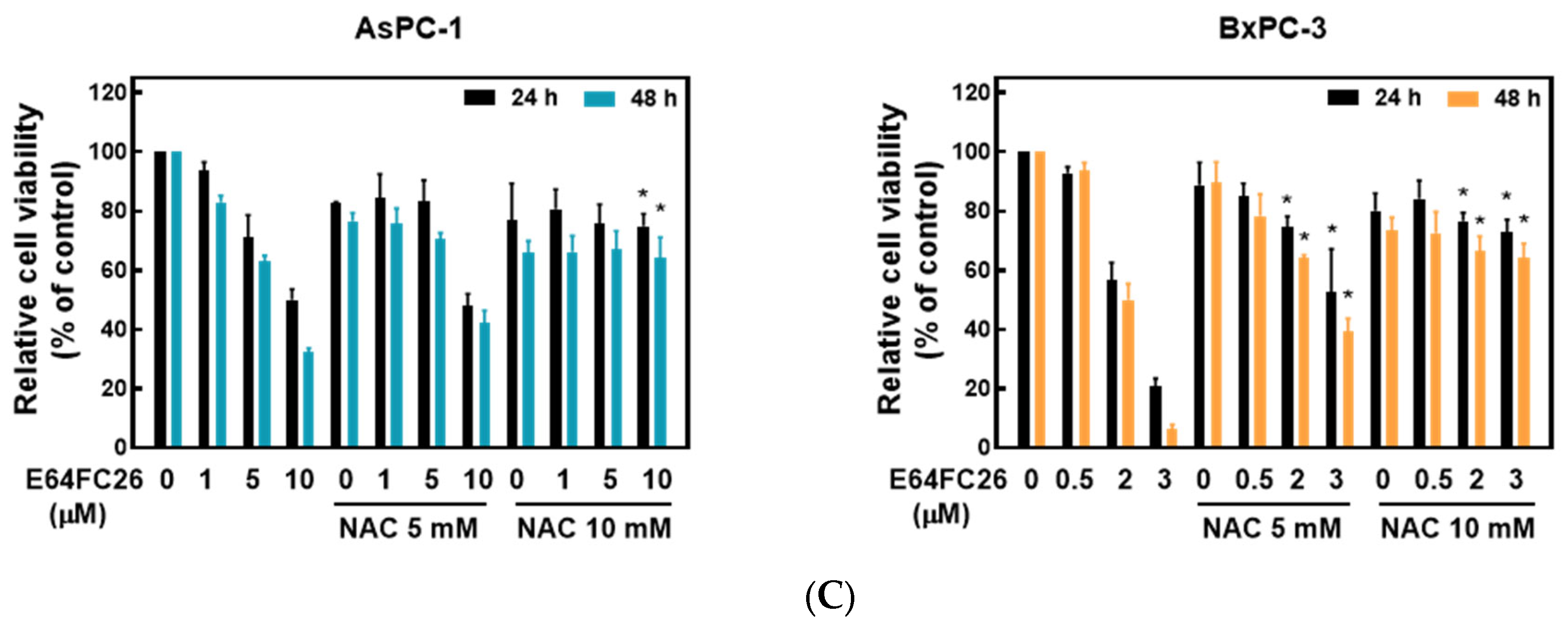
2.4. PDI Inhibitor E64FC26-Induced Cell Death Might Be Associated with Ferroptosis
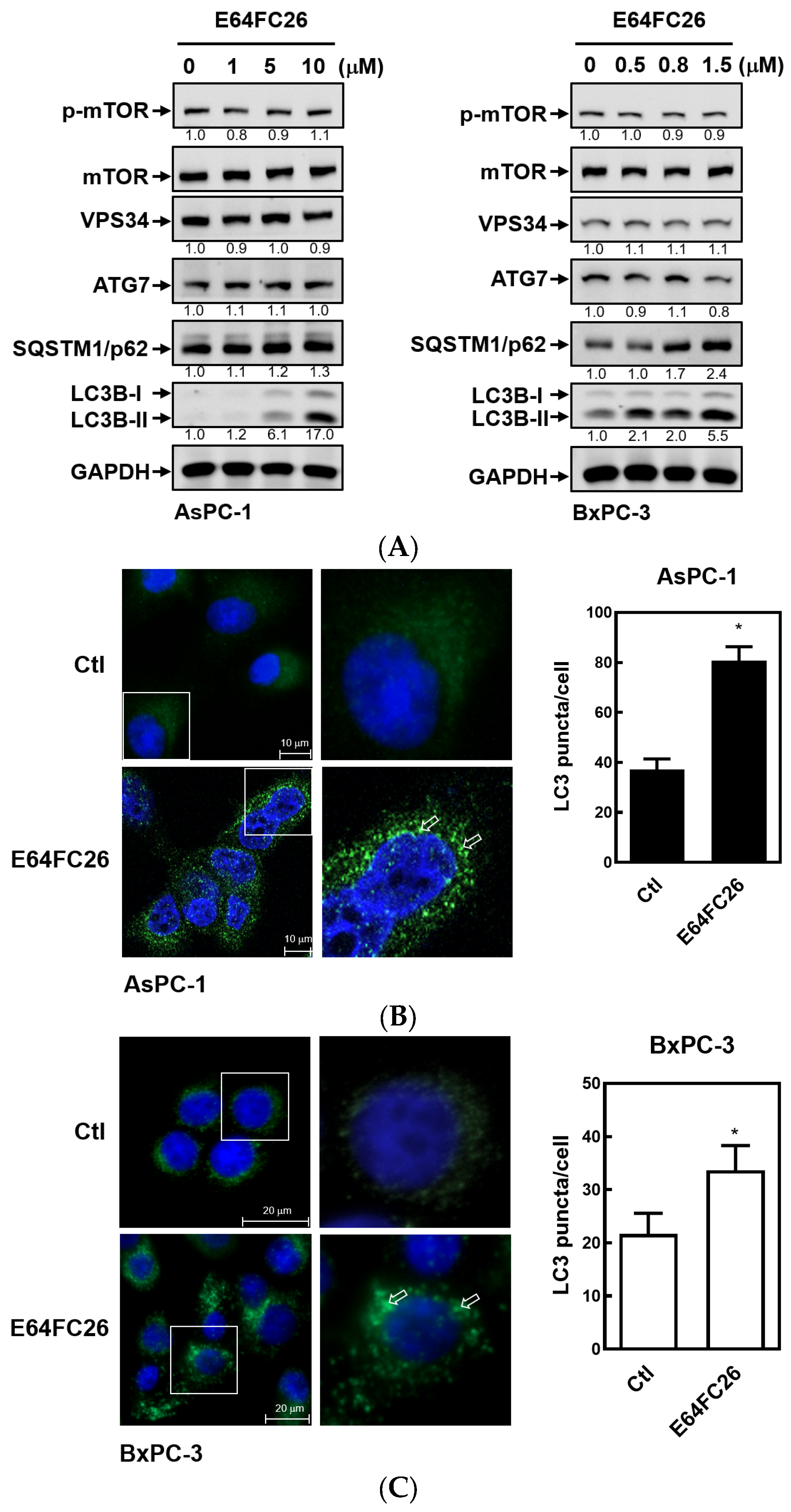
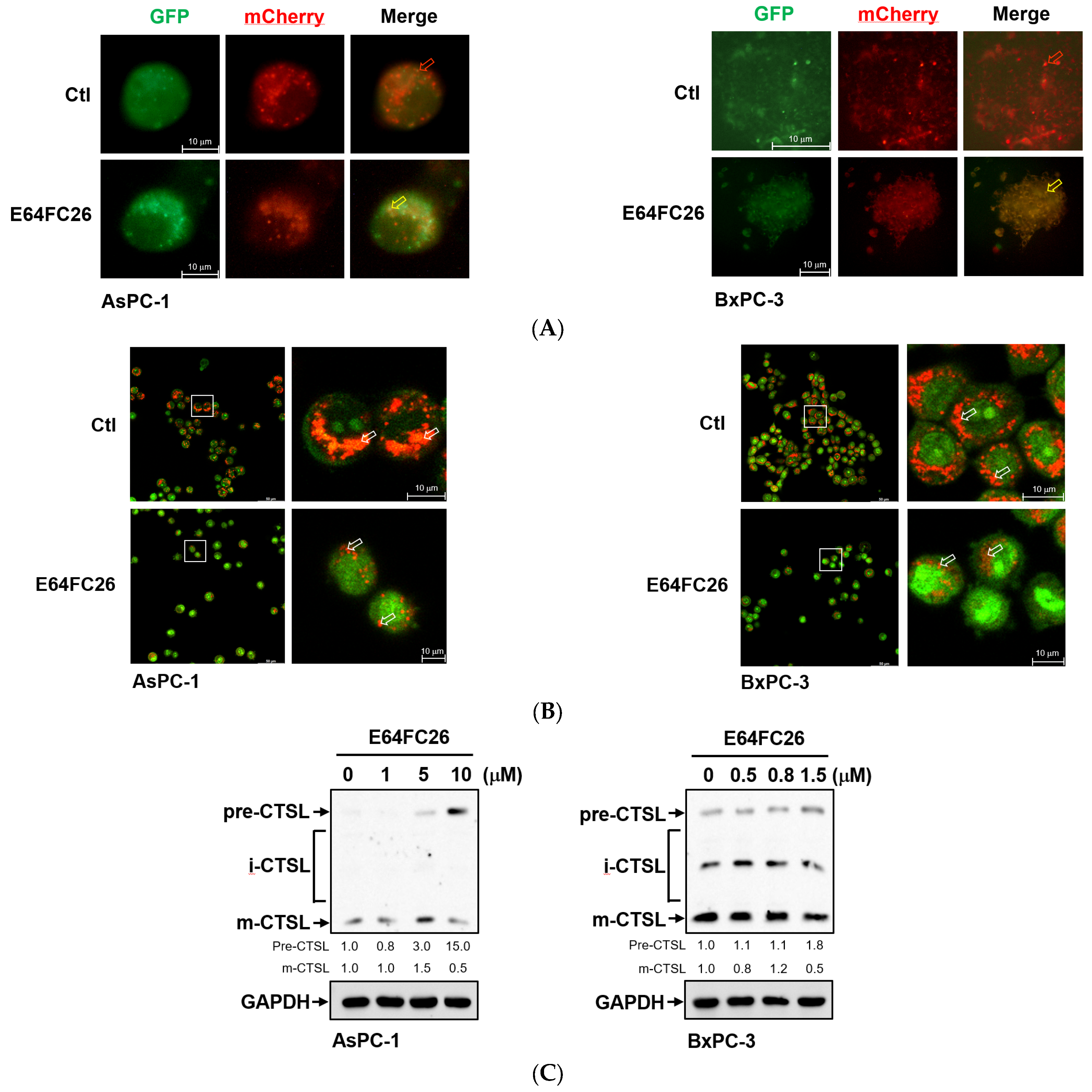
2.5. The PDI Inhibitor, E64FC26, Caused Autophagic Cell Death by Blocking Autolysosome Formation
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture
4.3. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay
4.4. Western Blot Analysis
4.5. Caspase Activity Assay
4.6. Transient Transfection
4.7. Acridine Orange (AO) Staining
4.8. Immunofluorescence (IF) Staining
4.9. Statistical Analysis
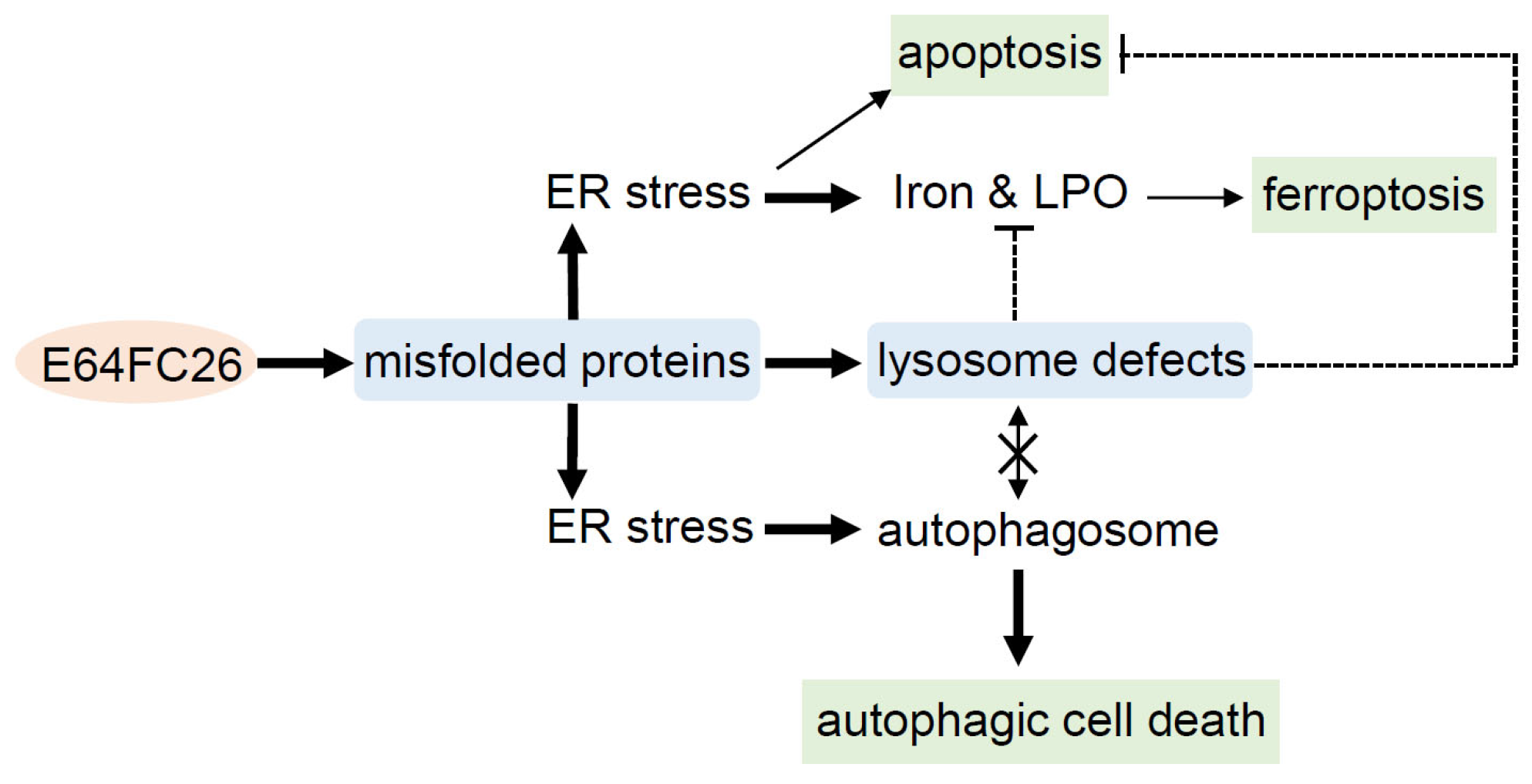
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lukas, J.; Pospech, J.; Oppermann, C.; Hund, C.; Iwanov, K.; Pantoom, S.; Petters, J.; Frech, M.; Seemann, S.; Thiel, F.G.; et al. Role of endoplasmic reticulum stress and protein misfolding in disorders of the liver and pancreas. Adv. Med. Sci. 2019, 64, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Bulleid, N.J.; Ellgaard, L. Multiple ways to make disulfides. Trends Biochem. Sci. 2011, 36, 485–492. [Google Scholar] [CrossRef]
- Shergalis, A.G.; Hu, S.; Bankhead, A., 3rd; Neamati, N. Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacol. Ther. 2020, 210, 107525. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chiu, J.; Chen, S.; Fang, C. Pathophysiological roles of cell surface and extracellular protein disulfide isomerase and their molecular mechanisms. Br. J. Pharmacol. 2021, 178, 2911–2930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Jha, V.; Kumari, T.; Manickam, V.; Assar, Z.; Olson, K.L.; Min, J.K.; Cho, J. ERO1-PDI Redox Signaling in Health and Disease. Antioxid. Redox Signal 2021, 35, 1093–1115. [Google Scholar] [CrossRef]
- Stepanchuk, A.A.; Stys, P.K. Amyloid dye pairs as spectral sensors for enhanced detection and differentiation of misfolded proteins. J. Photochem. Photobiol. B 2023, 248, 112786. [Google Scholar] [CrossRef]
- Yamada, Y.; Tomaru, U.; Ishizu, A.; Ito, T.; Kiuchi, T.; Ono, A.; Miyajima, S.; Nagai, K.; Higashi, T.; Matsuno, Y.; et al. Decreased proteasomal function accelerates cigarette smoke-induced pulmonary emphysema in mice. Lab. Investig. 2015, 95, 625–634. [Google Scholar] [CrossRef]
- Brusa, I.; Sondo, E.; Falchi, F.; Pedemonte, N.; Roberti, M.; Cavalli, A. Proteostasis Regulators in Cystic Fibrosis: Current Development and Future Perspectives. J. Med. Chem. 2022, 65, 5212–5243. [Google Scholar] [CrossRef]
- Ye, Z.W.; Zhang, J.; Aslam, M.; Blumental-Perry, A.; Tew, K.D.; Townsend, D.M. Protein disulfide isomerase family mediated redox regulation in cancer. Adv. Cancer Res. 2023, 160, 83–106. [Google Scholar] [CrossRef]
- Hoffstrom, B.G.; Kaplan, A.; Letso, R.; Schmid, R.S.; Turmel, G.J.; Lo, D.C.; Stockwell, B.R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Wise, R.; Duhachek-Muggy, S.; Qi, Y.; Zolkiewski, M.; Zolkiewska, A. Protein disulfide isomerases in the endoplasmic reticulum promote anchorage-independent growth of breast cancer cells. Breast Cancer Res. Treat. 2016, 157, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.S.; Serino, L.T.; Carvalho, C.M.; Lima, R.S.; Urban, C.A.; Cavalli, I.J.; Ribeiro, E.M. PDIA3 and PDIA6 gene expression as an aggressiveness marker in primary ductal breast cancer. Genet. Mol. Res. 2015, 14, 6960–6967. [Google Scholar] [CrossRef]
- Alavi, M.; Mah, V.; Maresh, E.L.; Bagryanova, L.; Horvath, S.; Chia, D.; Goodglick, L.; Liu, A.Y. High expression of AGR2 in lung cancer is predictive of poor survival. BMC Cancer 2015, 15, 655. [Google Scholar] [CrossRef]
- Mouawad, R.; Neamati, N. Inhibition of Protein Disulfide Isomerase (PDIA1) Leads to Proteasome-Mediated Degradation of Ubiquitin-like PHD and RING Finger Domain-Containing Protein 1 (UHRF1) and Increased Sensitivity of Glioblastoma Cells to Topoisomerase II Inhibitors. ACS Pharmacol. Transl. Sci. 2023, 6, 100–114. [Google Scholar] [CrossRef]
- Araujo, T.L.S.; Zeidler, J.D.; Oliveira, P.V.S.; Dias, M.H.; Armelin, H.A.; Laurindo, F.R.M. Protein disulfide isomerase externalization in endothelial cells follows classical and unconventional routes. Free Radic. Biol. Med. 2017, 103, 199–208. [Google Scholar] [CrossRef]
- Wolzak, K.; Vermunt, L.; Campo, M.D.; Jorge-Oliva, M.; van Ziel, A.M.; Li, K.W.; Smit, A.B.; Chen-Ploktkin, A.; Irwin, D.J.; Lemstra, A.W.; et al. Protein disulfide isomerases as CSF biomarkers for the neuronal response to tau pathology. Alzheimers Dement. 2023, 19, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Jang, I.; Pottekat, A.; Poothong, J.; Yong, J.; Lagunas-Acosta, J.; Charbono, A.; Chen, Z.; Scheuner, D.L.; Liu, M.; Itkin-Ansari, P.; et al. PDIA1/P4HB is required for efficient proinsulin maturation and ß cell health in response to diet induced obesity. eLife 2019, 8, e44528. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Butkevich, A.N.; Yamada, R.; Zhou, Y.; Debnath, B.; Duncan, R.; Zandi, E.; Petasis, N.A.; Neamati, N. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc. Natl. Acad. Sci. USA 2012, 109, 16348–16353. [Google Scholar] [CrossRef]
- Powell, L.E.; Foster, P.A. Protein disulphide isomerase inhibition as a potential cancer therapeutic strategy. Cancer Med. 2021, 10, 2812–2825. [Google Scholar] [CrossRef]
- Vatolin, S.; Phillips, J.G.; Jha, B.K.; Govindgari, S.; Hu, J.; Grabowski, D.; Parker, Y.; Lindner, D.J.; Zhong, F.; Distelhorst, C.W.; et al. Novel Protein Disulfide Isomerase Inhibitor with Anticancer Activity in Multiple Myeloma. Cancer Res. 2016, 76, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Reitz, A.B.; Manevich, Y.; McClure, J.J.; Champion, M.M.; Chou, C.J.; Sharik, M.E.; et al. Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia 2019, 33, 1011–1022. [Google Scholar] [CrossRef]
- Zhou, X.; Li, G.; Kaplan, A.; Gaschler, M.M.; Zhang, X.; Hou, Z.; Jiang, M.; Zott, R.; Cremers, S.; Stockwell, B.R.; et al. Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Kyani, A.; Tamura, S.; Yang, S.; Shergalis, A.; Samanta, S.; Kuang, Y.; Ljungman, M.; Neamati, N. Discovery and Mechanistic Elucidation of a Class of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma. ChemMedChem 2018, 13, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Yu, J.; Li, T.; Liu, Y.; Wang, X.; Zhang, J.; Wang, X.; Shi, G.; Lou, J.; Wang, L.; Wang, C.C.; et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020, 39, e103841. [Google Scholar] [CrossRef] [PubMed]
- Hurst, K.E.; Lawrence, K.A.; Reyes Angeles, L.; Ye, Z.; Zhang, J.; Townsend, D.M.; Dolloff, N.; Thaxton, J.E. Endoplasmic Reticulum Protein Disulfide Isomerase Shapes T Cell Efficacy for Adoptive Cellular Therapy of Tumors. Cells 2019, 8, 1514. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes. Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.S.A.; Zahari, S.; Syafruddin, S.E.; Firdaus-Raih, M.; Low, T.Y.; Mohtar, M.A. Functions and mechanisms of protein disulfide isomerase family in cancer emergence. Cell Biosci. 2022, 12, 129. [Google Scholar] [CrossRef] [PubMed]
- Khodier, C.; VerPlank, L.; Nag, P.P.; Pu, J.; Wurst, J.; Pilyugina, T.; Dockendorff, C.; Galinski, C.N.; Scalise, A.A.; Passam, F.; et al. Identification of ML359 as a Small Molecule Inhibitor of Protein Disulfide Isomerase. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Kaplan, A.; Gaschler, M.M.; Dunn, D.E.; Colligan, R.; Brown, L.M.; Palmer, A.G., 3rd; Lo, D.C.; Stockwell, B.R. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc. Natl. Acad. Sci. USA 2015, 112, E2245–E2252. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Gopal, S.; Sharda, A.; Passam, F.; Bowley, S.R.; Stopa, J.; Xue, G.; Yuan, C.; Furie, B.C.; Flaumenhaft, R.; et al. Quercetin-3-rutinoside Inhibits Protein Disulfide Isomerase by Binding to Its b’x Domain. J. Biol. Chem. 2015, 290, 23543–23552. [Google Scholar] [CrossRef]
- Cole, K.S.; Grandjean, J.M.D.; Chen, K.; Witt, C.H.; O’Day, J.; Shoulders, M.D.; Wiseman, R.L.; Weerapana, E. Characterization of an A-Site Selective Protein Disulfide Isomerase A1 Inhibitor. Biochemistry 2018, 57, 2035–2043. [Google Scholar] [CrossRef]
- Alfieri, M.L.; Cariola, A.; Panzella, L.; Napolitano, A.; d’Ischia, M.; Valgimigli, L.; Crescenzi, O. Disentangling the Puzzling Regiochemistry of Thiol Addition to o-Quinones. J. Org. Chem. 2022, 87, 4580–4589. [Google Scholar] [CrossRef]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Strobel, E.D.; Hyman, S.L.; Reitz, A.B.; Dolloff, N.G. Tuning isoform selectivity and bortezomib sensitivity with a new class of alkenyl indene PDI inhibitor. Eur. J. Med. Chem. 2020, 186, 111906. [Google Scholar] [CrossRef]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Sawai, H.; Funahashi, H.; Yamamoto, M.; Okada, Y.; Hayakawa, T.; Tanaka, M.; Takeyama, H.; Manabe, T. Interleukin-1alpha enhances integrin alpha(6)beta(1) expression and metastatic capability of human pancreatic cancer. Oncology 2003, 65, 167–173. [Google Scholar] [CrossRef]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef]
- Zhao, B.; Qin, C.; Li, Z.; Wang, Y.; Li, T.; Cao, H.; Yang, X.; Li, T.; Wang, W. Multidrug resistance genes screening of pancreatic ductal adenocarcinoma based on sensitivity profile to chemotherapeutic drugs. Cancer Cell Int. 2022, 22, 374. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J., 3rd; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef]
- Fu, X.; Cui, J.; Meng, X.; Jiang, P.; Zheng, Q.; Zhao, W.; Chen, X. Endoplasmic reticulum stress, cell death and tumor: Association between endoplasmic reticulum stress and the apoptosis pathway in tumors (Review). Oncol. Rep. 2021, 45, 801–808. [Google Scholar] [CrossRef]
- Kuang, H.; Sun, X.; Liu, Y.; Tang, M.; Wei, Y.; Shi, Y.; Li, R.; Xiao, G.; Kang, J.; Wang, F.; et al. Palmitic acid-induced ferroptosis via CD36 activates ER stress to break calcium-iron balance in colon cancer cells. FEBS J. 2023, 290, 3664–3687. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Park, Y.J.; Lee, S.J.; Lee, K.; Yoon, C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol. Lett. 2019, 303, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Bai, Y.; Jia, Y.; Zhao, Y.; Kang, R.; Tang, D.; Dai, E. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 2018, 503, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Sun, K.; Li, C.; Liao, S.; Yao, X.; Ouyang, Y.; Liu, Y.; Wang, Z.; Li, Z.; Yao, F. Ferritinophagy, a form of autophagic ferroptosis: New insights into cancer treatment. Front. Pharmacol. 2022, 13, 1043344. [Google Scholar] [CrossRef]
- Koksal, A.R.; Verne, G.N.; Zhou, Q. Endoplasmic reticulum stress in biological processing and disease. J. Investig. Med. 2021, 69, 309–315. [Google Scholar] [CrossRef]
- Yuan, W.; Fang, W.; Zhang, R.; Lyu, H.; Xiao, S.; Guo, D.; Ali, D.W.; Michalak, M.; Chen, X.Z.; Zhou, C.; et al. Therapeutic strategies targeting AMPK-dependent autophagy in cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119537. [Google Scholar] [CrossRef]
- Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D. Autophagy and Ferroptosis—What’s the Connection? Curr. Pathobiol. Rep. 2017, 5, 153–159. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Suk, F.M.; Chang, C.C.; Sun, P.C.; Ke, W.T.; Chung, C.C.; Lee, K.L.; Chan, T.S.; Liang, Y.C. MCPIP1 Enhances TNF-α-Mediated Apoptosis through Downregulation of the NF-κB/cFLIP Axis. Biology 2021, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lee, K.L.; Chan, T.S.; Chung, C.C.; Liang, Y.C. Histone Deacetylase Inhibitors Downregulate Calcium Pyrophosphate Crystal Formation in Human Articular Chondrocytes. Int. J. Mol. Sci. 2022, 23, 2604. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.C.; Chang, C.C.; Sheu, M.T.; Lin, S.Y.; Chung, C.C.; Teng, C.T.; Suk, F.M. The Antihistamine Deptropine Induces Hepatoma Cell Death through Blocking Autophagosome-Lysosome Fusion. Cancers 2020, 12, 1610. [Google Scholar] [CrossRef]
- Thomé, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, C.-S.; Lee, K.-L.; Huang, W.-J.; Su, F.-H.; Liang, Y.-C. Pan-Inhibition of Protein Disulfide Isomerase Caused Cell Death through Disrupting Cellular Proteostasis in Pancreatic Ductal Adenocarcinoma Cells. Int. J. Mol. Sci. 2023, 24, 16467. https://doi.org/10.3390/ijms242216467
Hung C-S, Lee K-L, Huang W-J, Su F-H, Liang Y-C. Pan-Inhibition of Protein Disulfide Isomerase Caused Cell Death through Disrupting Cellular Proteostasis in Pancreatic Ductal Adenocarcinoma Cells. International Journal of Molecular Sciences. 2023; 24(22):16467. https://doi.org/10.3390/ijms242216467
Chicago/Turabian StyleHung, Ching-Sheng, Kun-Lin Lee, Wei-Jan Huang, Fang-He Su, and Yu-Chih Liang. 2023. "Pan-Inhibition of Protein Disulfide Isomerase Caused Cell Death through Disrupting Cellular Proteostasis in Pancreatic Ductal Adenocarcinoma Cells" International Journal of Molecular Sciences 24, no. 22: 16467. https://doi.org/10.3390/ijms242216467
APA StyleHung, C.-S., Lee, K.-L., Huang, W.-J., Su, F.-H., & Liang, Y.-C. (2023). Pan-Inhibition of Protein Disulfide Isomerase Caused Cell Death through Disrupting Cellular Proteostasis in Pancreatic Ductal Adenocarcinoma Cells. International Journal of Molecular Sciences, 24(22), 16467. https://doi.org/10.3390/ijms242216467