
Cerebrospinal and Blood Biomarkers in Alzheimer’s Disease: Did Mild Cognitive Impairment Definition Affect Their Clinical Usefulness?



Abstract
:1. Background
2. Current AD Biomarkers: A Short Summary
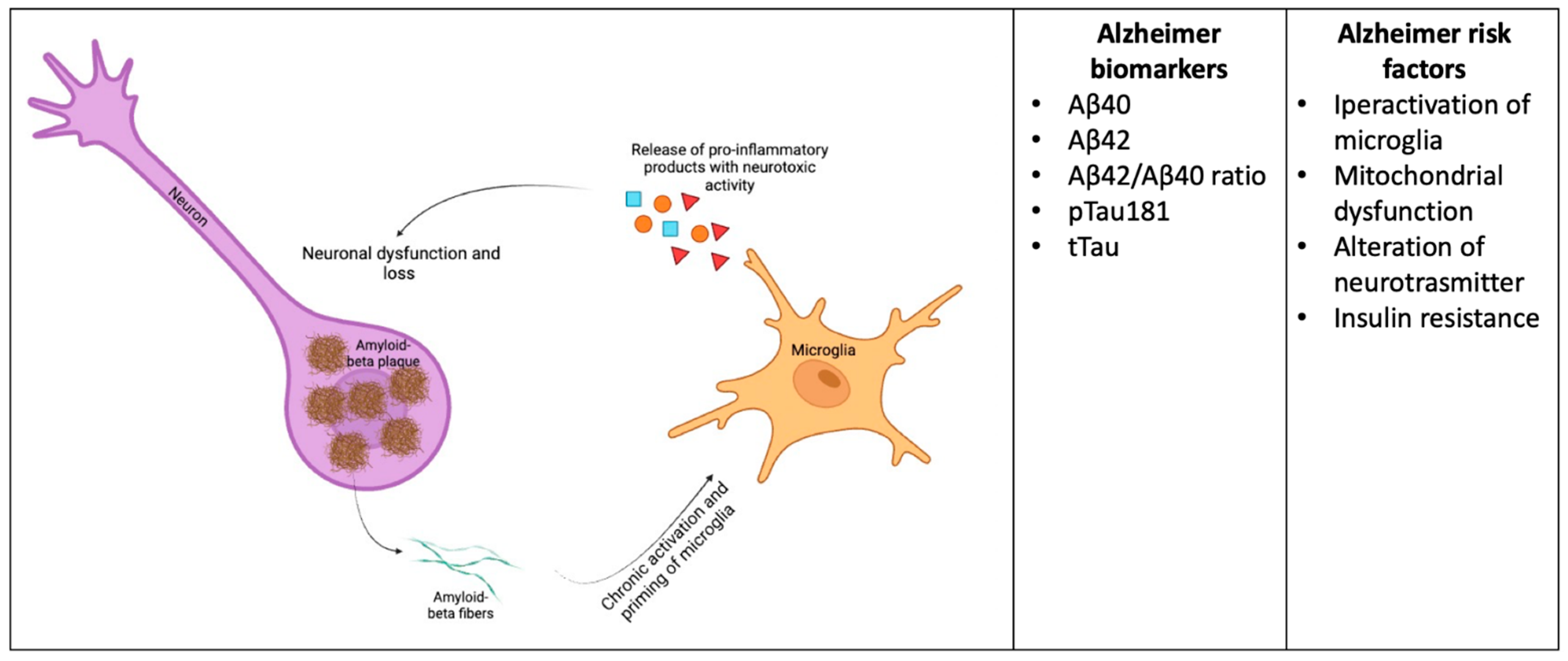
Microglia Biomarkers
3. Advantages and Flaws of AD Current Biomarkers
Flaws of the Studies on MCI and AD Biomarkers
4. Novel AD Biomarkers
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Alzheimer Europe. Dementia in Europe Yearbook 2019: Estimating the Prevalence of Dementia in Europe. 2020. Available online: https://www.alzheimer-europe.org/sites/default/files/alzheimer_europe_dementia_in_europe_yearbook_2019.pdf (accessed on 19 June 2023).
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, M. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.Y. γ-Secretase in Alzheimer’s disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Shen, W.; Su, J.; Cheng, B.; Li, D.; Liu, G.; Zhou, W.X.; Zhang, Y.X. Modulating the Balance of Synaptic and Extrasynaptic NMDA Receptors Shows Positive Effects against Amyloid-β-Induced Neurotoxicity. J. Alzheimers Dis. 2017, 57, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Shimada, H. APP Osaka Mutation in Familial Alzheimer’s Disease-Its Discovery, Phenotypes, and Mechanism of Recessive Inheritance. Int. J. Mol. Sci. 2020, 21, 1413. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, M.; Majmundar, L.; Sengupta, U.; Fung, L.; Kayed, R. Pathological tau signatures and nuclear alterations in neurons, astrocytes and microglia in Alzheimer’s disease, progressive supranuclear palsy, and dementia with Lewy bodies. Brain Pathol. 2023, 33, e13112. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef]
- Kitazawa, M.; Yamasaki, T.R.; LaFerla, F.M. Microglia as a potential bridge between the amyloid β-peptide and tau. Ann. N. Y. Acad. Sci. 2004, 1035, 85–103. [Google Scholar] [CrossRef]
- Adams, J.D. Probable Causes of Alzheimer’s Disease. Science 2021, 3, 16. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillod, M.C.; Dunne, B.; Haeberleinf, S.B.; Holtzmang, D.M.; Jagusth, W.; Jesseni, F.; Karlawishj, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Winblad, B.; O’Brien, J.T. Revised NIA-AA criteria for the diagnosis of Alzheimer’s disease: A step forward but not yet ready for widespread clinical use. Int. Psychogeriatr. 2011, 23, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, F.H.; Teunissen, C.E.; Scheltens, P.; Barkhof, F.; Frolich, L.; Kornhuber, J.; Wiltfang, J.; van Maurik, I.S.; Vos, S.J.; Bos, I.; et al. Biomarker-based prognosis for people with mild cognitive impairment (ABIDE): A modelling study. Lancet Neurol. 2019, 18, 1034–1044. [Google Scholar]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, D.S.; Ashton, N.J.; Blennow, K.; Zetterberg, H.; Simrén, J.; Lantero-Rodriguez, J.; Karikari, T.K.; Hiniker, A.; Rissman, R.A.; Salmon, D.P.; et al. Plasma biomarkers for Alzheimer’s Disease in relation to neuropathology and cognitive change. Acta Neuropathol. 2022, 143, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Stevenson-Hoare, J.; Heslegrave, A.; Leonenko, G.; Fathalla, D.; Bellou, E.; Luckcuck, L.; Marshall, R.; Sims, R.; Morgan, B.P.; Hardy, J.; et al. Plasma biomarkers and genetics in the diagnosis and prediction of Alzheimer’s disease. Brain 2023, 146, 690–699. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Fortea, J.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernández, S.; Altuna, M.; Pegueroles, J.; Montal, V.; et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet 2020, 395, 1988–1997. [Google Scholar] [CrossRef]
- Morenas-Rodríguez, E.; Li, Y.; Nuscher, B.; Franzmeier, N.; Xiong, C.; Suárez-Calvet, M.; Fagan, A.M.; Schultz, S.; Gordon, B.A.; Benzinger, T.L.S.; et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: A longitudinal observational study. Lancet Neurol. 2022, 21, 329–341. [Google Scholar] [CrossRef]
- Bivona, G.; Iemmolo, M.; Piccoli, T.; Agnello, L.; Lo Sasso, B.; Ciaccio, M.; Ghersi, G. High Cerebrospinal Fluid CX3CL1 Levels in Alzheimer’s Disease Patients but Not in Non-Alzheimer’s Disease Dementia. J. Clin. Med. 2022, 11, 5498. [Google Scholar] [CrossRef] [PubMed]
- Bivona, G.; Iemmolo, M.; Agnello, L.; Lo Sasso, B.; Gambino, C.M.; Giglio, R.V.; Scazzone, C.; Ghersi, G.; Ciaccio, M. Microglial Activation and Priming in Alzheimer’s Disease: State of the Art and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 884. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Finneran, D.J.; Nash, K.R. Neuroinflammation and fractalkine signaling in Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Zheng, H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J. Neurochem. 2016, 136, 475–491. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef]
- Bivona, G.; Iemmolo, M.; Ghersi, G. CX3CL1 Pathway as a Molecular Target for Treatment Strategies in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 8230. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Strosznajder, J.B. Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11677. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef]
- Kuhlmann, J.; Andreasson, U.; Pannee, J. CSF Abeta1–42—An excellent but complicated Alzheimer’s biomarker—A route to standardisation. Clin. Chim. Acta 2017, 467, 27–33. [Google Scholar] [CrossRef]
- Hok, A.H.Y.S.; Willemse, E.A.J.; Teunissen, C.E.; Del Campo, M. Guidelines for CSF processing and biobanking: Impact on the identification and development of optimal CSF protein biomarkers. Methods Mol. Biol. 2019, 2044, 27–50. [Google Scholar]
- Pannee, J.; Gobom, J.; Shaw, L.M. Round robin test on quantification of amyloid-beta 1–42 in cerebrospinal fluid by mass spectrometry. Alzheimers Dement. 2016, 12, 55–59. [Google Scholar] [CrossRef]
- Petersen, R.C. Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 2004, 256, 183–194. [Google Scholar] [CrossRef]
- Chatterjee, P.; Pedrini, S.; Ashton, N.J.; Tegg, M.; Goozee, K.; Singh, A.K.; Karikari, T.K.; Simrén, J.; Vanmechelen, E.; Armstrong, N.J.; et al. Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer’s disease. Alzheimers Dement. 2022, 18, 1141–1154. [Google Scholar] [CrossRef]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Tangalos, E.G.; Kokmen, E. Mild cognitive impairment: Clinical characterization and outcome. Arch. Neurol. 1999, 56, 303–308. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M. Towards defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging—Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimers Res. Ther. 2019, 11, 34. [Google Scholar] [CrossRef]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Andreasson, U.; Londos, E.; Minthon, L.; Blennow, K. Prediction of Alzheimer’s disease using the CSF Abeta42/ Abeta40 ratio in patients with mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2007, 23, 316–320. [Google Scholar] [CrossRef]
- Hertze, J.; Minthon, L.; Zetterberg, H.; Vanmechelen, E.; Blennow, K.; Hansson, O. Evaluation of CSF biomarkers as predictors of Alzheimer’s disease: A clinical follow-up study of 4.7 years. J. Alzheimers Dis. 2010, 21, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Chiasserini, D.; Eusebi, P.; Giannandrea, D.; Bellomo, G.; De Carlo, C.; Padiglioni, C.; Mastrocola, S.; Lisetti, V.; Calabresi, P. Performance of Aβ1-40, Aβ1-42, total tau, and phosphorylated tau as predictors of dementia in a cohort of patients with mild cognitive impairment. J. Alzheimers Dis. 2012, 29, 229–238. [Google Scholar] [CrossRef]
- Frölich, L.; Peters, O.; Lewczuk, P.; Gruber, O.; Teipel, S.J.; Gertz, H.J.; Jahn, H.; Jessen, F.; Kurz, A.; Luckhaus, C.; et al. Incremental value of biomarker combinations to predict progression of mild cognitive impairment to Alzheimer’s dementia. Alzheimers Res. Ther. 2017, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Baldeiras, I.; Silva-Spínola, A.; Lima, M.; Leitão, M.J.; Durães, J.; Vieira, D.; Tábuas-Pereira, M.; Cruz, V.T.; Rocha, R.; Alves, L.; et al. Alzheimer’s Disease Diagnosis Based on the Amyloid, Tau, and Neurodegeneration Scheme (ATN) in a Real-Life Multicenter Cohort of General Neurological Centers. J. Alzheimers Dis. 2022, 90, 419–432. [Google Scholar] [CrossRef]
- Karikari, T.K.; Ashton, N.J.; Rodriguez, J.L.; Schöll, M.; Höglund, K.; Brinkmalm, G.; Zetterberg, H.; Blennow, K.A.; Pascoal, C.T.; Benedet, A.L.; et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020, 19, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef]
- Chen, T.B.; Lee, Y.J.; Lin, S.Y.; Chen, J.P.; Hu, C.J.; Wang, P.N.; Cheng, I.H. Plasma Aβ42 and total tau predict cognitive decline in amnestic mild cognitive impairment. Sci. Rep. 2019, 9, 13984. [Google Scholar] [CrossRef]
- Mahaman, Y.A.R.; Embaye, K.S.; Huang, F.; Li, L.; Zhu, F.; Wang, J.Z.; Liu, R.; Feng, J.; Wang, X. Biomarkers used in Alzheimer’s disease diagnosis, treatment, and prevention. Ageing Res. Rev. 2022, 74, 101544. [Google Scholar] [CrossRef]
- Carlyle, B.C.; Trombetta, B.A.; Arnold, S.E. Proteomic Approaches for the Discovery of Biofluid Biomarkers of Neurodegenerative Dementias. Proteomes 2018, 6, 32. [Google Scholar] [CrossRef]
- Janelidze, S.; Berron, D.; Smith, R.; Strindberg, O.; Proctor, N.K.; Dage, J.L.; Stomrud, E.; Palmqvist, S.; Mattsson-Carlgren, N.; Hansson, O. Associations of plasma Phospho-Tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA Neurol. 2021, 78, 149–156. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Petersen, M.; Johnson, L.; O’Bryant, S.E. Plasma Total Tau and Neurobehavioral Symptoms of Cognitive Decline in Cognitively Normal Older Adults. Front. Psychol. 2021, 12, 774049. [Google Scholar] [CrossRef]
- Pase, M.P.; Beiser, A.S.; Himali, J.J.; Satizabal, C.L.; Aparicio, H.J.; DeCarli, C.; Chêne, G.; Dufouil, C.; Seshadri, S. Assessment of plasma total tau level as a predictive biomarker for dementia and related endophenotypes. JAMA Neurol. 2019, 76, 598–606. [Google Scholar] [CrossRef]
- Salvadó, G.; Larsson, V.; Cody, K.A.; Cullen, N.C.; Jonaitis, E.M.; Stomrud, E.; Kollmorgen, G.; Wild, N.; Palmqvist, S.; Janelidze, S. Optimal combinations of CSF biomarkers for predicting cognitive decline and clinical conversion in cognitively unimpaired participants and mild cognitive impairment patients: A multi-cohort study. Alzheimers Dement. 2023, 19, 2943–2955. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, Y.; Li, T.; Cai, Y.; Han, Y. Amyloid-β as a Blood Biomarker for Alzheimer’s Disease: A Review of Recent Literature. J. Alzheimers Dis. 2020, 73, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C. Biomarkers of Alzheimer disease in plasma. NeuroRx 2004, 1, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [PubMed]
- Verberk, I.M.W.; Thijssen, E.; Koelewijn, J.; Mauroo, K.; Vanbrabant, J.; de Wilde, A.; Zwan, M.D.; Verfaillie, S.C.J.; Ossenkoppele, R.; Barkhof, F.; et al. Combination of plasma amyloid beta(1-42/1-40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res. Ther. 2020, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Verberk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; Del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mielke, M.M. An update on blood-based markers of Alzheimer’s disease using the SiMoA platform. Neurol. Ther. 2019, 8, 73–82. [Google Scholar] [CrossRef]
- Jia, L.; Qiua, Q.; Zhanga, H.; Chub, L.; Duc, Y.; Zhangd, J.; Zhoue, C.; Liangf, F.; Shig, S.; Wanget, S.; et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019, 15, 1071–1080. [Google Scholar] [CrossRef]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef]
- Johansson, C.; Thordardottir, S.; Laffita-Mesa, J.; Rodriguez-Vieitez, E.; Zetterberg, H.; Blennow, K.; Graff, C. Plasma biomarker profiles in autosomal dominant Alzheimer’s disease. Brain 2023, 146, 1132–1140. [Google Scholar] [CrossRef]
- Yoong, S.Q.; Lu, J.; Sing, H.; Gyanwali, B.; Tan, Y.Q.; Wu, X.V. The prognostic utility of CSF neurogranin in predicting future cognitive decline in the Alzheimer’s disease continuum: A systematic review and meta-analysis with narrative synthesis. Aging Res. Rev. 2021, 72, 101491. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, S.J.; Teunissen, C.E.; Pool, R.; Shipley, M.J.; Teumer, A.; Chouraki, V.; Melo van Lent, D.; Tynkkynen, J.; Fischer, K.; Hernesniemi, J.; et al. Circulating metabolites and general cognitive ability and dementia: Evidence from 11 cohort studies. Alzheimers Dement. 2018, 14, 707–722. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Biomarkers | Advantages | Flaws |
|---|---|---|
| Classical CSF biomarkers (CSF Aβ40, Aβ42, Aβ42/Aβ40 ratio, pTau181 and tTau) | Standardization process; quality program for harmonization; strong correlation with pathomechanisms | Invasive; poor repeatability; relatively small specificity (pTau and Tau) |
| Microglia biomarkers | Strong correlation with pathomechanisms | No validation studies; poor specificity |
| Blood classical biomarkers (plasma Aβ40, Aβ42, Aβ42/Aβ40 ratio, pTau181 and tTau) | Easy to perform; repeatability; not invasive | No validation studies; no clear clinical usefulness |
| Blood novel biomarker (NfL, neuronal-derived exosomes, and neuronal-enriched extracellular vesicles) | Repeatability; not invasive | Poor evidence on their clinical usefulness; no specificity; further studies required |
| Author | Cohort Characteristics | Sample Size | Main Findings | Year | Ref. |
|---|---|---|---|---|---|
| Hansson et al. | Stable MCI, pre-AD MCI | 137 | Abeta42/Abeta40 ratio is a predictive biomarker for AD | 2007 | [46] |
| Hertze et al. | Stable MCI and AD | 260 | Aβ42 and Tau have low predictive value for AD diagnosis | 2010 | [47] |
| Frolich et al. | Stable MCI, pre-AD MCI, and AD | 115 | Combination of Aβ42/Aβ40, pTau, and tTau has best value for AD than tTau alone and hippocampal volume | 2017 | [49] |
| Baldeiras et al. | AD and non-AD based on ATN scheme | 1128 | Aβ-related markers are the best predictors of AD | 2022 | [50] |
| Chatterjee et al. | Cognitively unimpaired subjects | 100 | Plasma GFAP, pTau181, pTau231, and NFL correlate with cognition, but not with hippocampal volume. | 2022 | [41] |
| Karikari | AD continuum | 1131 | pTau predicts AD along the AD continuum | 2020 | [51] |
| Janelidze | AD, MCI and cognitively unimpaired patients | 589 | Plasma Tau predicts AD in cognitively unimpaired patients | 2020 | [52] |
| Chen | MCI | 22 | Aβ42 and Tau are good predictors of AD development in MCI subjects | 2019 | [53] |
| Smirnov | MCI, dementia | 312 | plasma PTau181 and 231 are good predictors of AD | 2022 | [14] |
| Bateman | Autosomal AD mutation carriers | 128 | CSF Aβ42 and Tau decline 25 and 15 years before symptom onset, respectively. | 2012 | [21] |
| Stevenson-Hoare | AD | 1439 | Aβ40/Aβ42 ratio, GFAP, and NfL display the best prediction accuracy for AD diagnosis | 2023 | [15] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bivona, G.; Iemmolo, M.; Ghersi, G. Cerebrospinal and Blood Biomarkers in Alzheimer’s Disease: Did Mild Cognitive Impairment Definition Affect Their Clinical Usefulness? Int. J. Mol. Sci. 2023, 24, 16908. https://doi.org/10.3390/ijms242316908
Bivona G, Iemmolo M, Ghersi G. Cerebrospinal and Blood Biomarkers in Alzheimer’s Disease: Did Mild Cognitive Impairment Definition Affect Their Clinical Usefulness? International Journal of Molecular Sciences. 2023; 24(23):16908. https://doi.org/10.3390/ijms242316908
Chicago/Turabian StyleBivona, Giulia, Matilda Iemmolo, and Giulio Ghersi. 2023. "Cerebrospinal and Blood Biomarkers in Alzheimer’s Disease: Did Mild Cognitive Impairment Definition Affect Their Clinical Usefulness?" International Journal of Molecular Sciences 24, no. 23: 16908. https://doi.org/10.3390/ijms242316908
APA StyleBivona, G., Iemmolo, M., & Ghersi, G. (2023). Cerebrospinal and Blood Biomarkers in Alzheimer’s Disease: Did Mild Cognitive Impairment Definition Affect Their Clinical Usefulness? International Journal of Molecular Sciences, 24(23), 16908. https://doi.org/10.3390/ijms242316908