
A Biomimetic Multiparametric Assay to Characterise Anti-Amyloid Drugs
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
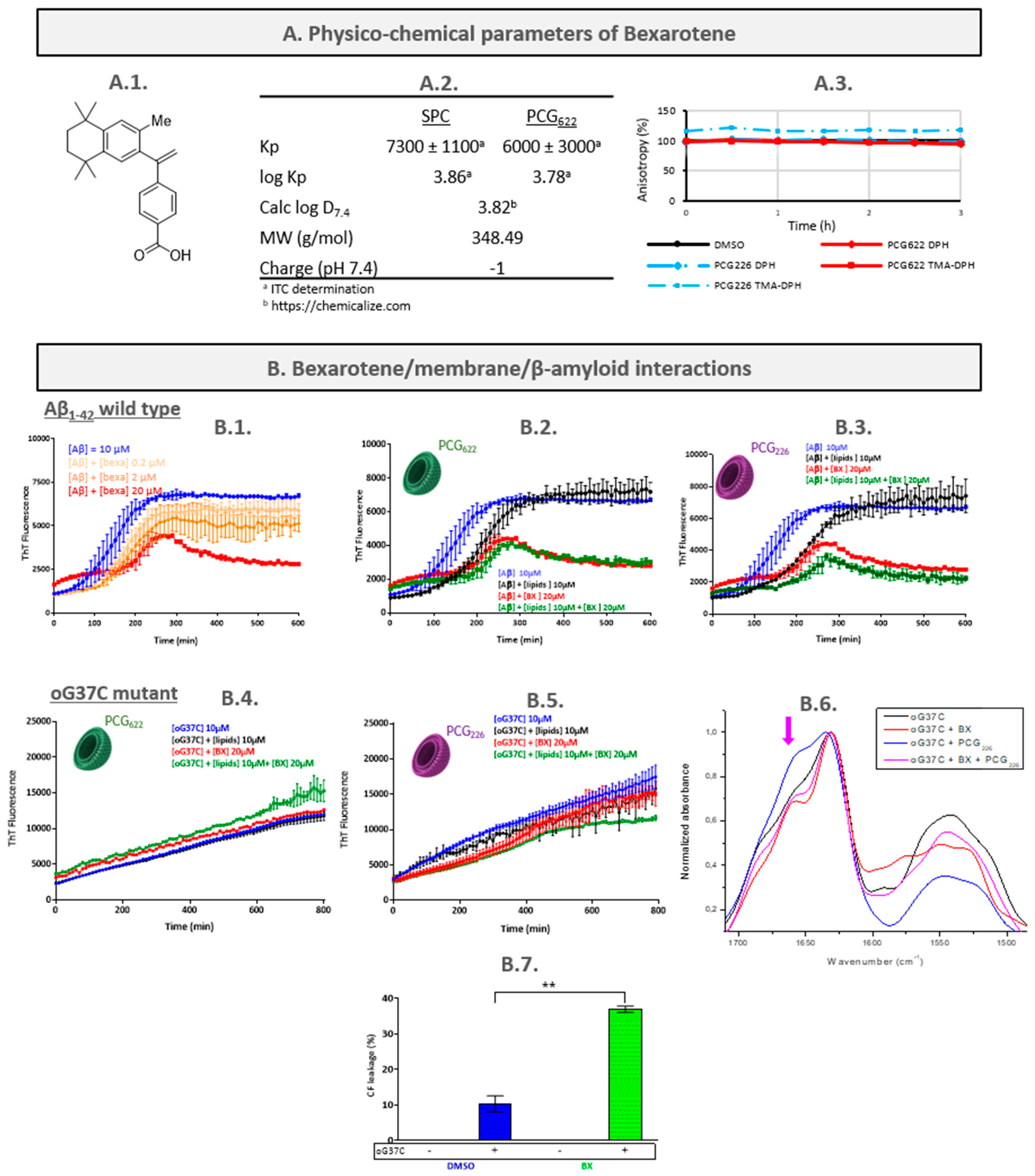
2.1. Bexarotene (BX)
2.1.1. Characterisation of the Bexarotene/LUVs Interactions
- Determination of bexarotene partition coefficient
- Assessment of bexarotene/membranes interactions by fluorescence anisotropy measurement (FPA)
2.1.2. Characterisation of the Bexarotene/Peptide Interactions
2.1.3. Characterisation of the Peptide/Bexarotene/LUVs Interactions
- Aβ1-42/bexarotene/LUVs interactions
- oG37C/bexarotene/LUVs interactions
- BX mechanism hypothesis and discussion
2.2. Chicago Sky Blue 6B (CSB)
2.2.1. Characterisation of the CSB/LUVs Interactions
- Determination of CSB partition coefficient
- Assessment of CSB/membranes interactions by fluorescence anisotropy measurement (FPA)
2.2.2. Characterisation of the CSB/Peptide Interactions
2.2.3. Characterisation of the Peptide/CSB/LUVs Interactions
- Aβ1-42/CSB/LUVs interactions
- oG37C/CSB/LUVs interactions
- CSB mechanism hypothesis and discussion
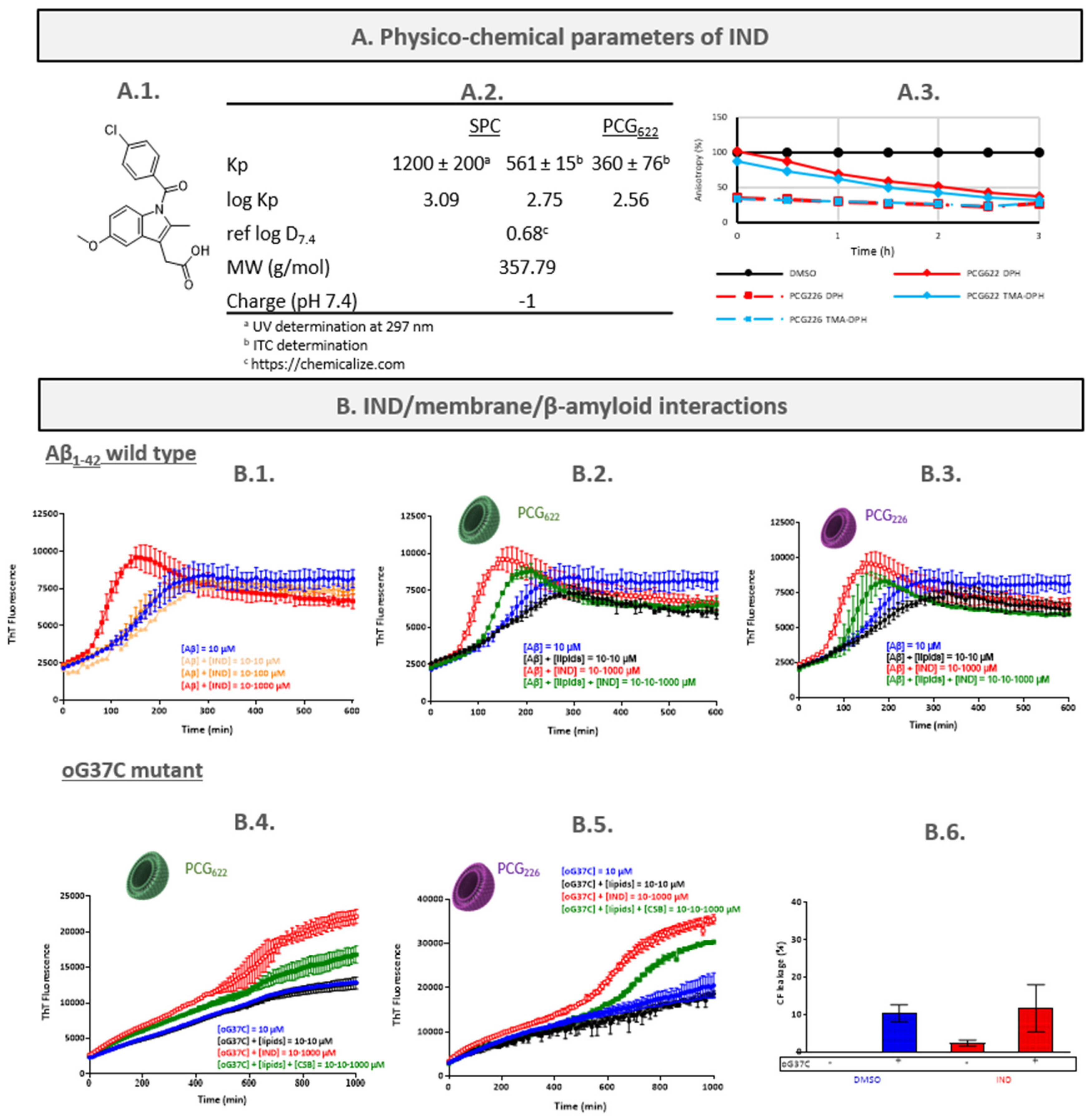
2.3. Indomethacin (IND)
2.3.1. Characterisation of the IND/LUVs Interactions
- Determination of IND partition coefficient
- Assessment of IND/membranes interactions by fluorescence anisotropy measurement (FPA)
2.3.2. Characterisation of the IND/Peptide Interactions
2.3.3. Characterisation of the Peptide/IND/LUVs Interactions
- Aβ1-42/IND/LUVs interactions
- oG37C/IND/LUVs interactions
- IND mechanism hypothesis and discussion
3. Materials and Methods
3.1. Materials
3.2. Production and Purification of oG37C
3.3. Liposomes
3.3.1. Formulation of Liposomes
3.3.2. Characterisation of Liposomes by Dynamic Light Scattering
3.4. Lipid Quantification by NMR Spectroscopy
3.5. Determination of Partition Coefficient by Derivative UV-Spectrophotometry
3.6. Determination of Partition Coefficient by Isothermal Titration Calorimetry (ITC)
3.7. Fluorescence Polarisation Anisotropy Experiments
3.8. Aβ Peptides Aggregation Kinetic Assay (ThT, Thioflavin T Fluorescence)
3.9. Liposomes Leakage Assay (LLA)
3.10. Attenuated Total Reflectance-Fourier Transform InfraRed Spectroscopy (ATR-FTIR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ghiso, J.; Frangione, B. Amyloidosis and Alzheimer’s Disease. Adv. Drug Deliv. Rev. 2002, 54, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- 2022 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2022, 18, 700–789. [CrossRef] [PubMed]
- Weglinski, C.; Jeans, A. Amyloid-β in Alzheimer’s Disease–Front and Centre after All? Neuronal Signal. 2023, 7, NS20220086. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Heneka, M.T. Truncated and Modified Amyloid-Beta Species. Alzheimers Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Rauk, A. The Chemistry of Alzheimer’s Disease. Chem. Soc. Rev. 2009, 38, 2698–2715. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Chakrabarti, M.; Govindaraju, T. Function and Toxicity of Amyloid Beta and Recent Therapeutic Interventions Targeting Amyloid Beta in Alzheimer’s Disease. Chem. Commun. 2015, 51, 13434–13450. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; La Rosa, C.; Milardi, D. Amyloid-Mediated Mechanisms of Membrane Disruption. Biophysica 2021, 1, 137–156. [Google Scholar] [CrossRef]
- Lee, M.-C.; Yu, W.-C.; Shih, Y.-H.; Chen, C.-Y.; Guo, Z.-H.; Huang, S.-J.; Chan, J.C.C.; Chen, Y.-R. Zinc Ion Rapidly Induces Toxic, off-Pathway Amyloid-β Oligomers Distinct from Amyloid-β Derived Diffusible Ligands in Alzheimer’s Disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef]
- Muschol, M.; Hoyer, W. Amyloid Oligomers as On-Pathway Precursors or off-Pathway Competitors of Fibrils. Front. Mol. Biosci. 2023, 10, 1120416. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Gao, F.; Hu, M.; Wang, X. Recent Developments in the Chemical Biology of Amyloid-β Oligomer Targeting. Org. Biomol. Chem. 2023, 21, 4540–4552. [Google Scholar] [CrossRef]
- Vignaud, H.; Bobo, C.; Lascu, I.; Sörgjerd, K.M.; Zako, T.; Maeda, M.; Salin, B.; Lecomte, S.; Cullin, C. A Structure-Toxicity Study of Aß42 Reveals a New Anti-Parallel Aggregation Pathway. PLoS ONE 2013, 8, e80262. [Google Scholar] [CrossRef]
- Smeralda, W.; Since, M.; Cardin, J.; Corvaisier, S.; Lecomte, S.; Cullin, C.; Malzert-Fréon, A. β-Amyloid Peptide Interactions with Biomimetic Membranes: A Multiparametric Characterization. Int. J. Biol. Macromol. 2021, 181, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Chaibva, M.; Gao, X.; Jain, P.; Campbell, W.A.; Frey, S.L.; Legleiter, J. Sphingomyelin and GM1 Influence Huntingtin Binding to, Disruption of, and Aggregation on Lipid Membranes. ACS Omega 2018, 3, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Chia, S.; Galvagnion, C.; Michaels, T.C.T.; Bellaiche, M.M.J.; Ruggeri, F.S.; Sanguanini, M.; Idini, I.; Kumita, J.R.; Sparr, E.; et al. Cholesterol Catalyses Aβ42 Aggregation through a Heterogeneous Nucleation Pathway in the Presence of Lipid Membranes. Nat. Chem. 2018, 10, 673–683. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, M.; Fusco, G.; De Simone, A. Membrane Interactions and Toxicity by Misfolded Protein Oligomers. Front. Cell Dev. Biol. 2021, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dey, S.; Bhowmik, D.; Maiti, S. Coexisting Ordered and Disordered Membrane Phases Have Distinct Modes of Interaction with Disease-Associated Oligomers. J. Phys. Chem. B 2022, 126, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Attar, A.; Rahimi, F.; Bitan, G. Modulators of Amyloid Protein Aggregation and Toxicity: EGCG and CLR01. Transl. Neurosci. 2013, 4, 385–409. [Google Scholar] [CrossRef]
- Jourdan, J.-P.; Since, M.; El Kihel, L.; Lecoutey, C.; Corvaisier, S.; Legay, R.; Sopkova-de Oliveira Santos, J.; Cresteil, T.; Malzert-Fréon, A.; Rochais, C.; et al. Novel Benzylidenephenylpyrrolizinones with Pleiotropic Activities Potentially Useful in Alzheimer’s Disease Treatment. Eur. J. Med. Chem. 2016, 114, 365–379. [Google Scholar] [CrossRef]
- Jourdan, J.-P.; Since, M.; El Kihel, L.; Lecoutey, C.; Corvaisier, S.; Legay, R.; Sopková-de Oliveira Santos, J.; Cresteil, T.; Malzert-Fréon, A.; Rochais, C.; et al. Benzylphenylpyrrolizinones with Anti-Amyloid and Radical Scavenging Effects, Potentially Useful in Alzheimer’s Disease Treatment. ChemMedChem 2017, 12, 913–916. [Google Scholar] [CrossRef]
- Habchi, J.; Chia, S.; Limbocker, R.; Mannini, B.; Ahn, M.; Perni, M.; Hansson, O.; Arosio, P.; Kumita, J.R.; Challa, P.K.; et al. Systematic Development of Small Molecules to Inhibit Specific Microscopic Steps of Aβ42 Aggregation in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2017, 114, E200–E208. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine Enhances Aβ42 Aggregation but Suppresses Its Toxicity by Displacing Oligomers from Cell Membranes. Nat. Commun. 2019, 10, 225. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular Mechanism of Thioflavin-T Binding to Amyloid Fibrils. Biochim. Biophys. Acta BBA-Proteins Proteom. 2010, 1804, 1405–1412. [Google Scholar] [CrossRef]
- Sarroukh, R.; Goormaghtigh, E.; Ruysschaert, J.-M.; Raussens, V. ATR-FTIR: A “Rejuvenated” Tool to Investigate Amyloid Proteins. Biochim. Biophys. Acta BBA-Biomembr. 2013, 1828, 2328–2338. [Google Scholar] [CrossRef]
- Berthelot, K.; Cullin, C.; Lecomte, S. What Does Make an Amyloid Toxic: Morphology, Structure or Interaction with Membrane? Biochimie 2013, 95, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Henry, S.; Vignaud, H.; Bobo, C.; Decossas, M.; Lambert, O.; Harte, E.; Alves, I.D.; Cullin, C.; Lecomte, S. Interaction of Aβ 1–42 Amyloids with Lipids Promotes “Off-Pathway” Oligomerization and Membrane Damage. Biomacromolecules 2015, 16, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Smeralda, W.; Since, M.; Corvaisier, S.; Legay, R.; Voisin-Chiret, A.-S.; Malzert-Freon, A. Microplate Assay for Lipophilicity Determination Using Intrinsic Fluorescence of Drugs: Application to a Promising Anticancer Lead, Pyridoclax. Eur. J. Pharm. Sci. 2019, 131, 75–83. [Google Scholar] [CrossRef]
- Magalhães, L.M.; Nunes, C.; Lúcio, M.; Segundo, M.A.; Reis, S.; Lima, J.L.F.C. High-Throughput Microplate Assay for the Determination of Drug Partition Coefficients. Nat. Protoc. 2010, 5, 1823–1830. [Google Scholar] [CrossRef]
- Nunes, C.; Lopes, D.; Pinheiro, M.; Pereira-Leite, C.; Reis, S. In Vitro Assessment of NSAIDs-Membrane Interactions: Significance for Pharmacological Actions. Pharm. Res. 2013, 30, 2097–2107. [Google Scholar] [CrossRef]
- Loureiro, D.R.P.; Soares, J.X.; Lopes, D.; Macedo, T.; Yordanova, D.; Jakobtorweihen, S.; Nunes, C.; Reis, S.; Pinto, M.M.M.; Afonso, C.M.M. Accessing Lipophilicity of Drugs with Biomimetic Models: A Comparative Study Using Liposomes and Micelles. Eur. J. Pharm. Sci. 2018, 115, 369–380. [Google Scholar] [CrossRef]
- Boehm, M.F.; Zhang, L.; Zhi, L.; McClurg, M.R.; Berger, E.; Wagoner, M.; Mais, D.E.; Suto, C.M.; Davies, P.J.A.; Heyman, R.A.; et al. Design and Synthesis of Potent Retinoid X Receptor Selective Ligands That Induce Apoptosis in Leukemia Cells. J. Med. Chem. 1995, 38, 3146–3155. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Di Scala, C.; Yahi, N.; Troadec, J.-D.; Sadelli, K.; Chahinian, H.; Garmy, N. Bexarotene Blocks Calcium-Permeable Ion Channels Formed by Neurotoxic Alzheimer’s β-Amyloid Peptides. ACS Chem. Neurosci. 2014, 5, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-Directed Therapeutics Rapidly Clear -Amyloid and Reverse Deficits in AD Mouse Models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef]
- Cummings, J.L.; Zhong, K.; Kinney, J.W.; Heaney, C.; Moll-Tudla, J.; Joshi, A.; Pontecorvo, M.; Devous, M.; Tang, A.; Bena, J. Double-Blind, Placebo-Controlled, Proof-of-Concept Trial of Bexarotene in Moderate Alzheimer’s Disease. Alzheimer’s Res. Ther. 2016, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Arosio, P.; Perni, M.; Costa, A.R.; Yagi-Utsumi, M.; Joshi, P.; Chia, S.; Cohen, S.I.A.; Müller, M.B.D.; Linse, S.; et al. An Anticancer Drug Suppresses the Primary Nucleation Reaction That Initiates the Production of the Toxic Aβ42 Aggregates Linked with Alzheimer’s Disease. Sci. Adv. 2016, 2, e1501244. [Google Scholar] [CrossRef] [PubMed]
- Huy, P.D.Q.; Thai, N.Q.; Bednarikova, Z.; Phuc, L.H.; Linh, H.Q.; Gazova, Z.; Li, M.S. Bexarotene Does Not Clear Amyloid Beta Plaques but Delays Fibril Growth: Molecular Mechanisms. ACS Chem. Neurosci. 2017, 8, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Scheidt, H.A.; Winkler, E.; Basset, G.; Heinel, H.; Hutchison, J.M.; LaPointe, L.M.; Sanders, C.R.; Steiner, H.; Huster, D. Bexarotene Binds to the Amyloid Precursor Protein Transmembrane Domain, Alters Its α-Helical Conformation, and Inhibits γ-Secretase Nonselectively in Liposomes. ACS Chem. Neurosci. 2018, 9, 1702–1713. [Google Scholar] [CrossRef]
- Baddireddy, K.; Poojary, S. A Novel Contrast Stain for the Rapid Diagnosis of Dermatophytoses: A Cross-Sectional Comparative Study of Chicago Sky Blue 6B Stain, Potassium Hydroxide Mount and Culture. Indian J. Dermatol. 2019, 64, 311–314. [Google Scholar] [CrossRef]
- Beirith, A.; Santos, A.R.S.; Calixto, J.B. Mechanisms Underlying the Nociception and Paw Oedema Caused by Injection of Glutamate into the Mouse Paw. Brain Res. 2002, 924, 219–228. [Google Scholar] [CrossRef]
- He, Z.; Yan, L.; Yong, Z.; Dong, Z.; Dong, H.; Gong, Z. Chicago Sky Blue 6B, a Vesicular Glutamate Transporters Inhibitor, Attenuates Methamphetamine-Induced Hyperactivity and Behavioral Sensitization in Mice. Behav. Brain Res. 2013, 239, 172–176. [Google Scholar] [CrossRef]
- Min, J.-O.; Strohäker, T.; Jeong, B.-C.; Zweckstetter, M.; Lee, S.-J. Chicago Sky Blue 6B Inhibits α-Synuclein Aggregation and Propagation. Mol. Brain 2022, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small Molecule Inhibitors of Aggregation Indicate That Amyloid β Oligomerization and Fibrillization Pathways Are Independent and Distinct. J. Biol. Chem. 2007, 282, 10311–10324. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S. The Pharmacology of Indomethacin: Headache. Headache J. Head Face Pain 2016, 56, 436–446. [Google Scholar] [CrossRef] [PubMed]
- de Jong, D.; Jansen, R.; Hoefnagels, W.; Jellesma-Eggenkamp, M.; Verbeek, M.; Borm, G.; Kremer, B. No Effect of One-Year Treatment with Indomethacin on Alzheimer’s Disease Progression: A Randomized Controlled Trial. PLoS ONE 2008, 3, e1475. [Google Scholar] [CrossRef]
- Nunes, C.; Brezesinski, G.; Lopes, D.; Lima, J.L.F.C.; Reis, S.; Lúcio, M. Lipid–Drug Interaction: Biophysical Effects of Tolmetin on Membrane Mimetic Systems of Different Dimensionality. J. Phys. Chem. B 2011, 115, 12615–12623. [Google Scholar] [CrossRef]
- Brittes, J.; Lúcio, M.; Nunes, C.; Lima, J.L.F.C.; Reis, S. Effects of Resveratrol on Membrane Biophysical Properties: Relevance for Its Pharmacological Effects. Chem. Phys. Lipids 2010, 163, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, V.; Dean, D.N.; Rana, P.; Vaidya, A.; Ghosh, P. Cause and Consequence of Aβ–Lipid Interactions in Alzheimer Disease Pathogenesis. Biochim. Biophys. Acta BBA-Biomembr. 2018, 1860, 1652–1662. [Google Scholar] [CrossRef]
- Šarić, A.; Chebaro, Y.C.; Knowles, T.P.; Frenkel, D. Crucial Role of Nonspecific Interactions in Amyloid Nucleation. Proc. Natl. Acad. Sci. USA 2014, 111, 17869–17874. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural Conversion of Neurotoxic Amyloid-Β1–42 Oligomers to Fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef]
- Agopian, A.; Guo, Z. Structural Origin of Polymorphism of Alzheimer’s Amyloid β-Fibrils. Biochem. J. 2012, 447, 43–50. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Bolognesi, M.L.; Cavalli, A.; Melchiorre, C.; Andrisano, V. Insight Into the Kinetic of Amyloid β (1–42) Peptide Self-Aggregation: Elucidation of Inhibitors’ Mechanism of Action. ChemBioChem 2007, 8, 2152–2161. [Google Scholar] [CrossRef]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of Amyloid-Β42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]
- Gu, L.; Tran, J.; Jiang, L.; Guo, Z. A New Structural Model of Alzheimer’s Aβ42 Fibrils Based on Electron Paramagnetic Resonance Data and Rosetta Modeling. J. Struct. Biol. 2016, 194, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.; Villar-Alvarez, E.; Taboada, P.; Rocha, F.A.; Damas, A.M.; Martins, P.M. What Can the Kinetics of Amyloid Fibril Formation Tell about Off-Pathway Aggregation? J. Biol. Chem. 2016, 291, 2018–2032. [Google Scholar] [CrossRef] [PubMed]
- Tousi, B. The Emerging Role of Bexarotene in the Treatment of Alzheimer’s Disease: Current Evidence. Neuropsychiatr. Dis. Treat. 2015, 11, 311. [Google Scholar] [CrossRef] [PubMed]
- Cizas, P.; Budvytyte, R.; Morkuniene, R.; Moldovan, R.; Broccio, M.; Lösche, M.; Niaura, G.; Valincius, G.; Borutaite, V. Size-Dependent Neurotoxicity of β-Amyloid Oligomers. Arch. Biochem. Biophys. 2010, 496, 84–92. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Bobo, C.; Chaignepain, S.; Henry, S.; Vignaud, H.; Améadan, A.; Marchal, C.; Prado, E.; Doutch, J.; Schmitter, J.-M.; Nardin, C.; et al. Synthetic Toxic Aβ 1–42 Oligomers Can Assemble in Different Morphologies. Biochim. Biophys. Acta BBA-Gen. Subj. 2017, 1861, 1168–1176. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Blumenthal, R.; Klausner, R.D. Carboxyfluorescein Leakage Assay for Lipoprotein-Liposome Interaction. Methods Enzymol. 1986, 128, 657–668. [Google Scholar] [CrossRef]
- Ghosal, K.; Haag, M.; Verghese, P.B.; West, T.; Veenstra, T.; Braunstein, J.B.; Bateman, R.J.; Holtzman, D.M.; Landreth, G.E. A Randomized Controlled Study to Evaluate the Effect of Bexarotene on Amyloid-β and Apolipoprotein E Metabolism in Healthy Subjects. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 110–120. [Google Scholar] [CrossRef]
- Pollack, S.J.; Hawtin, S.R.; Tailor, V.J.; Shearman, M.S. Sulfonated Dyes Attenuate the Toxic Effects β-Amyloid in a Structure-Specific Fashion. Neurosci. Lett. 1995, 197, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Roseth, S.; Fykse, E.M.; Fonnum, F. Uptake of L-Glutamate into Rat Brain Synaptic Vesicles: Effect of Inhibitors That Bind Specifically to the Glutamate Transporter. J. Neurochem. 2002, 65, 96–103. [Google Scholar] [CrossRef]
- Nunes, C.; Brezesinski, G.; Pereira-Leite, C.; Lima, J.L.F.C.; Reis, S.; Lúcio, M. NSAIDs Interactions with Membranes: A Biophysical Approach. Langmuir 2011, 27, 10847–10858. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Plowman, S.J.; Lichtenberger, L.M.; Hancock, J.F. The Anti-Inflammatory Drug Indomethacin Alters Nanoclustering in Synthetic and Cell Plasma Membranes. J. Biol. Chem. 2010, 285, 35188–35195. [Google Scholar] [CrossRef] [PubMed]
- Fearon, A.D.; Stokes, G.Y. Thermodynamics of Indomethacin Adsorption to Phospholipid Membranes. J. Phys. Chem. B 2017, 121, 10508–10518. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Serpell, L.C. Membrane and Surface Interactions of Alzheimer’s Aβ Peptide-Insights into the Mechanism of Cytotoxicity: Membrane Interactions of Alzheimer’s Aβ Peptide. FEBS J. 2011, 278, 3905–3917. [Google Scholar] [CrossRef]
- Ewald, M.; Henry, S.; Lambert, E.; Feuillie, C.; Bobo, C.; Cullin, C.; Lecomte, S.; Molinari, M. High Speed Atomic Force Microscopy to Investigate the Interactions between Toxic Aβ 1–42 Peptides and Model Membranes in Real Time: Impact of the Membrane Composition. Nanoscale 2019, 11, 7229–7238. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of Univalent Ions across the Lamellae of Swollen Phospholipids. J. Mol. Biol. 1965, 13, 238–252, IN26–IN27. [Google Scholar] [CrossRef]
- Hein, R.; Uzundal, C.B.; Hennig, A. Simple and Rapid Quantification of Phospholipids for Supramolecular Membrane Transport Assays. Org. Biomol. Chem. 2016, 14, 2182–2185. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Jimah, J.; Schlesinger, P.; Tolia, N. Liposome Disruption Assay to Examine Lytic Properties of Biomolecules. BIO-Protocol 2017, 7, e2433. [Google Scholar] [CrossRef]
- Matos, C.; Lima, J.L.C.; Reis, S.; Lopes, A.; Bastos, M. Interaction of Antiinflammatory Drugs with EPC Liposomes: Calorimetric Study in a Broad Concentration Range. Biophys. J. 2004, 86, 946–954. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smeralda, W.; Since, M.; Corvaisier, S.; Fayolle, D.; Cardin, J.; Duprey, S.; Jourdan, J.-P.; Cullin, C.; Malzert-Freon, A. A Biomimetic Multiparametric Assay to Characterise Anti-Amyloid Drugs. Int. J. Mol. Sci. 2023, 24, 16982. https://doi.org/10.3390/ijms242316982
Smeralda W, Since M, Corvaisier S, Fayolle D, Cardin J, Duprey S, Jourdan J-P, Cullin C, Malzert-Freon A. A Biomimetic Multiparametric Assay to Characterise Anti-Amyloid Drugs. International Journal of Molecular Sciences. 2023; 24(23):16982. https://doi.org/10.3390/ijms242316982
Chicago/Turabian StyleSmeralda, Willy, Marc Since, Sophie Corvaisier, Dimitri Fayolle, Julien Cardin, Sylvain Duprey, Jean-Pierre Jourdan, Christophe Cullin, and Aurélie Malzert-Freon. 2023. "A Biomimetic Multiparametric Assay to Characterise Anti-Amyloid Drugs" International Journal of Molecular Sciences 24, no. 23: 16982. https://doi.org/10.3390/ijms242316982
APA StyleSmeralda, W., Since, M., Corvaisier, S., Fayolle, D., Cardin, J., Duprey, S., Jourdan, J.-P., Cullin, C., & Malzert-Freon, A. (2023). A Biomimetic Multiparametric Assay to Characterise Anti-Amyloid Drugs. International Journal of Molecular Sciences, 24(23), 16982. https://doi.org/10.3390/ijms242316982