
Expanding Genotype–Phenotype Correlation of CLCNKA and CLCNKB Variants Linked to Hearing Loss
 , and
, and
Abstract
:1. Introduction
2. Results
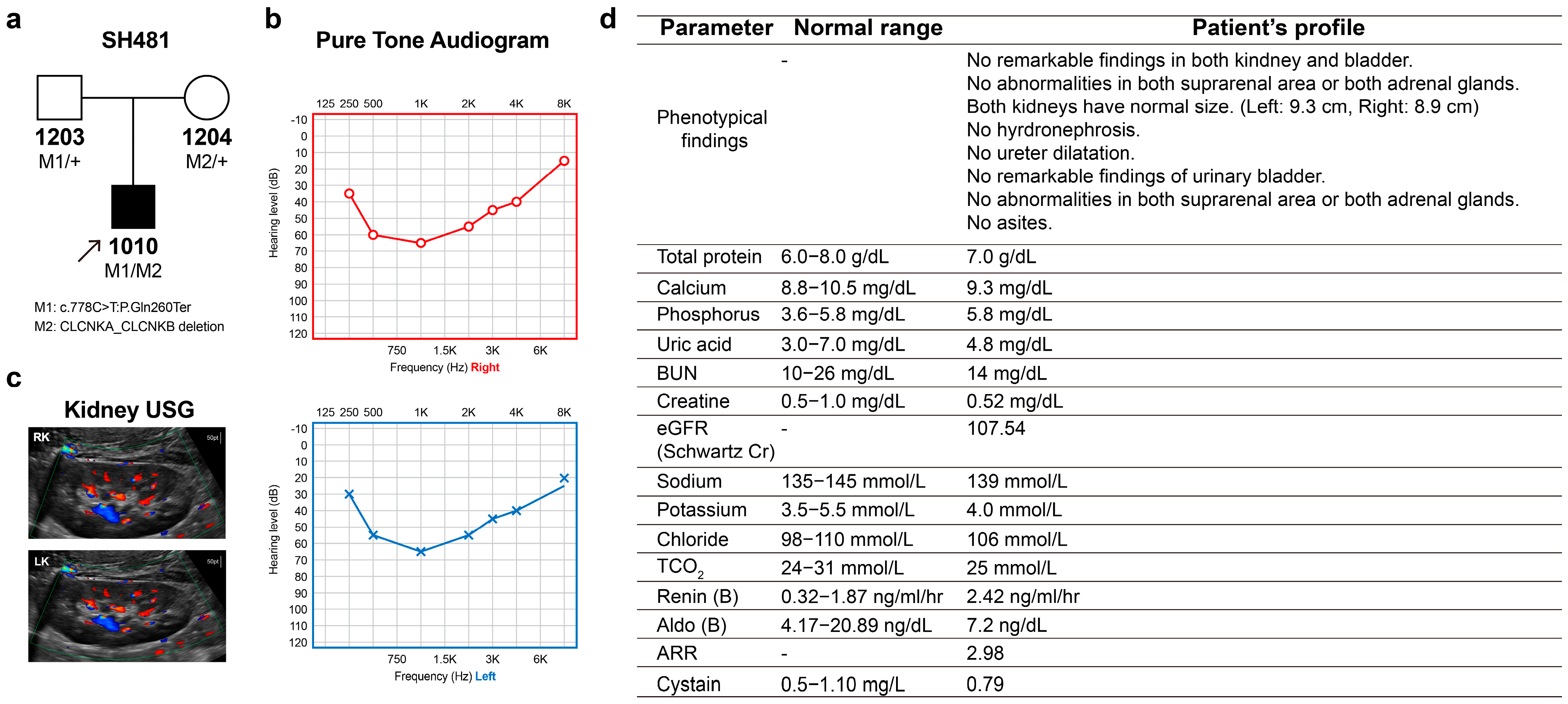
2.1. Phenotype
2.2. Exome Sequencing and Data Analysis
2.3. Whole Genome Sequencing and Data Analysis
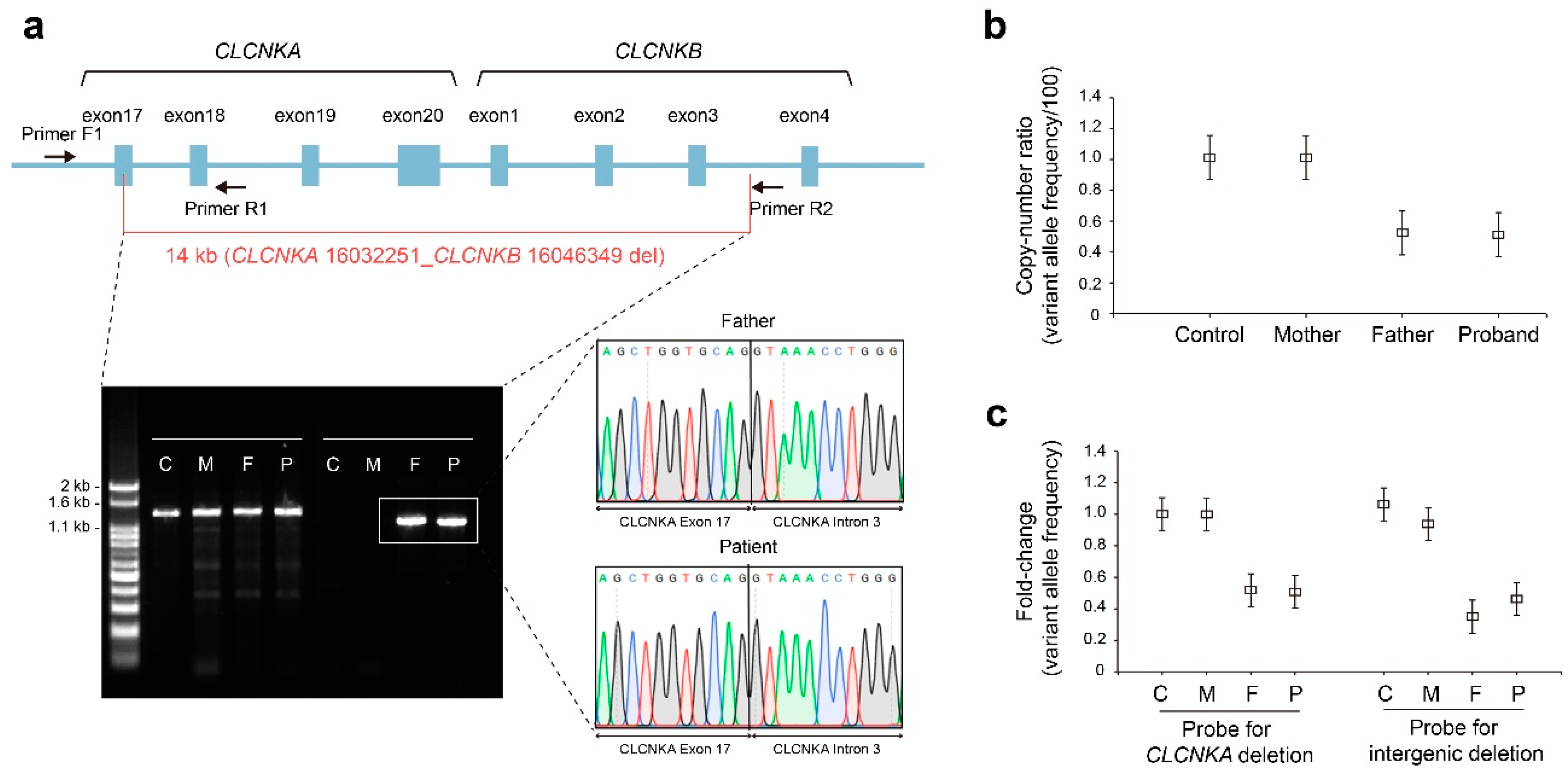
2.4. Molecular Genetic Analysis
2.5. Phenotype–Genotype Correlation
3. Discussion
4. Materials and Methods
4.1. Participants and Clinical Assessment
4.2. Exome Sequencing and Bioinformatic Analysis
4.3. Whole-Genome Sequencing and Bioinformatic Analysis
4.4. Breakpoint PCR, Digital Droplet PCR, and Sanger Sequencing
4.5. Laboratory Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SNHL | Sensorineural hearing loss |
| WGS | Whole genome sequencing |
| MET | Sensory mechanoelectrical transduction |
| BS | Bartter syndrome |
| ACMG-AMP | American College of Medical Genetics and Genomics/Association for Molecular Pathology |
| SVs | Structure variations |
| ddPCR | Digital-droplet PCR |
| VAF | Variant allele frequency |
| CNV | Copy number variations |
| gnomAD | Gemone Aggregation Database |
| GRCh38 | Human reference genome |
| GATK | Genome analysis toolkit |
| SNV | Single nucleotide variations |
| MAFs | Minor allele frequencies |
| KOVA2 | Korean Variant Archive for a reference database of genetic variations in the Korean population |
| CADD | Combined Annotation Dependent Depletion |
| REVEL | Rare Exome Variant Ensemble Learner |
| IGV | Integrative genomics viewer |
| USG | Ultrasonography |
References
- Fahlke, C.; Fischer, M. Physiology and pathophysiology of ClC-K/barttin channels. Front. Physiol. 2010, 1, 155. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Uchida, S.; Ito, H.; Hata, M.; Hiroe, M.; Marumo, F.; Sasaki, S. Two isoforms of a chloride channel predominantly expressed in thick ascending limb of Henle’s loop and collecting ducts of rat kidney. J. Biol. Chem. 1994, 269, 17677–17683. [Google Scholar] [CrossRef] [PubMed]
- Krämer, B.K.; Bergler, T.; Stoelcker, B.; Waldegger, S. Mechanisms of Disease: The kidney-specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat. Clin. Pract. Nephrol. 2008, 4, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Andrini, O.; Eladari, D.; Picard, N. ClC-K Kidney Chloride Channels: From Structure to Pathology. Handb. Exp. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Hennings, J.C.; Andrini, O.; Picard, N.; Paulais, M.; Huebner, A.K.; Cayuqueo, I.K.; Bignon, Y.; Keck, M.; Cornière, N.; Böhm, D.; et al. The ClC-K2 Chloride Channel Is Critical for Salt Handling in the Distal Nephron. J. Am. Soc. Nephrol. 2017, 28, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Rickheit, G.; Maier, H.; Strenzke, N.; Andreescu, C.E.; De Zeeuw, C.I.; Muenscher, A.; A Zdebik, A.; Jentsch, T.J. Endocochlear potential depends on Cl− channels: Mechanism underlying deafness in Bartter syndrome IV. EMBO J. 2008, 27, 2907–2917. [Google Scholar] [CrossRef]
- Patuzzi, R. Ion flow in stria vascularis and the production and regulation of cochlear endolymph and the endolymphatic potential. Hearth Res. 2011, 277, 4–19. [Google Scholar] [CrossRef]
- Estevez, R.; Boettger, T.; Stein, V.; Birkenhager, R.; Otto, E.; Hildebrandt, F.; Jentsch, T.J. Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 2001, 414, 558–561. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Konrad, M.; Jeck, N.; Waldegger, P.; Reinalter, S.C.; Holder, M.; Seyberth, H.W.; Waldegger, S. Salt Wasting and Deafness Resulting from Mutations in Two Chloride Channels. N. Engl. J. Med. 2004, 350, 1314–1319. [Google Scholar] [CrossRef]
- Cunha, T.D.S.; Heilberg, I.P. Bartter syndrome: Causes, diagnosis, and treatment. Int. J. Nephrol. Renovasc. Dis. 2018, 11, 291–301. [Google Scholar] [CrossRef]
- Simon, D.B.; Bindra, R.S.; Mansfield, T.A.; Nelson-Williams, C.; Mendonca, E.; Stone, R.; Schurman, S.; Nayir, A.; Alpay, H.; Bakkaloglu, A.; et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 1997, 17, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Nozu, K.; Inagaki, T.; Fu, X.J.; Nozu, Y.; Kaito, H.; Kanda, K.; Sekine, T.; Igarashi, T.; Nakanishi, K.; Yoshikawa, N.; et al. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J. Med. Genet. 2007, 45, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Birkenhäger, R.; Otto, E.; Schürmann, M.J.; Vollmer, M.; Ruf, E.-M.; Maier-Lutz, I.; Beekmann, F.; Fekete, A.; Omran, H.; Feldmann, D.; et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 2001, 29, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.; Anwar, S.; Fischer, M.; Ahmed, Z.M.; Khan, S.Y.; Janssen, A.G.; Zafar, A.U.; Scholl, U.; Husnain, T.; Belyantseva, I.A.; et al. Molecular Basis of DFNB73: Mutations of BSND Can Cause Nonsyndromic Deafness or Bartter Syndrome. Am. J. Hum. Genet. 2009, 85, 273–280. [Google Scholar] [CrossRef]
- Shafique, S.; Siddiqi, S.; Schraders, M.; Oostrik, J.; Ayub, H.; Bilal, A.; Ajmal, M.; Seco, C.Z.; Strom, T.M.; Mansoor, A.; et al. Genetic Spectrum of Autosomal Recessive Non-Syndromic Hearing Loss in Pakistani Families. PLoS ONE 2014, 9, e100146. [Google Scholar] [CrossRef]
- Coppola, M.A.; Pusch, M.; Imbrici, P.; Liantonio, A. Small Molecules Targeting Kidney ClC-K Chloride Channels: Applications in Rare Tubulopathies and Common Cardiovascular Diseases. Biomolecules 2023, 13, 710. [Google Scholar] [CrossRef] [PubMed]
- Koster, A.K.; Wood, C.A.P.; Thomas-Tran, R.; Chavan, T.S.; Almqvist, J.; Choi, K.-H.; Du Bois, J.; Maduke, M. A selective class of inhibitors for the CLC-Ka chloride ion channel. Proc. Natl. Acad. Sci. USA 2018, 115, E4900–E4909. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Matsumura, Y.; Uchida, S.; Kondo, Y.; Miyazaki, H.; Ko, S.B.; Hayama, A.; Morimoto, T.; Liu, W.; Arisawa, M.; Sasaki, S.; et al. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat. Genet. 1999, 21, 95–98. [Google Scholar] [CrossRef]
- Robitaille, P.; Merouani, A.; He, N.; Pei, Y. Bartter syndrome in two sisters with a novel mutation of the CLCNKB gene, one with deafness. Eur. J. Pediatr. 2011, 170, 1209–1211. [Google Scholar] [CrossRef]
- Ansar, M.; Lee, K.; Naqvi, S.K.-U.; Andrade, P.B.; Basit, S.; Santos-Cortez, R.L.P.; Ahmad, W.; Leal, S.M. A new autosomal recessive nonsyndromic hearing impairment locus DFNB96 on chromosome 1p36.31–p36.13. J. Hum. Genet. 2011, 56, 866–868. [Google Scholar] [CrossRef]
- Zheng, H.; Xu, J.; Wang, Y.; Lin, Y.; Hu, Q.; Li, X.; Chu, J.; Sun, C.; Chai, Y.; Pang, X. Identification and Characterization of a Cryptic Genomic Deletion-Insertion in EYA1 Associated with Branchio-Otic Syndrome. Neural Plast. 2021, 2021, 5524381. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Nawate, M.; Takahashi, Y.; Mizoguchi, Y.; Sugihara, S.; Yoshimoto, M.; Murakami, M.; Adachi, M.; Tachibana, K.; Mochizuki, H.; et al. Molecular Analysis of the CLCNKB Gene in Japanese Patients with Classic Bartter Syndrome. Endocr. J. 2006, 53, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Lupski, J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002, 18, 74–82. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Camps, C.; Giacopuzzi, E.; Taylor, J.M.; Hashim, M.; Calpena, E.; Kaisaki, P.J.; Hashimoto, A.; Yu, J.; Sanders, E.; et al. Structural and non-coding variants increase the diagnostic yield of clinical whole genome sequencing for rare diseases. Genome Med. 2023, 15, 94. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.; Pagnamenta, A.T.; Mack, H.G.; Savige, J.; Giacopuzzi, E.; Lines, K.E.; Taylor, J.C.; Thakker, R.V. The Bartter-Gitelman Spectrum: 50-Year Follow-up with Revision of Diagnosis After Whole-Genome Sequencing. J. Endocr. Soc. 2022, 6, bvac079. [Google Scholar] [CrossRef]
- Choy, K.W. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. N. Engl. J. Med. 2019, 380, 200–201. [Google Scholar]
- Stölting, G.; Fischer, M.; Fahlke, C. CLC channel function and dysfunction in health and disease. Front. Physiol. 2014, 5, 378. [Google Scholar] [CrossRef]
- Hayama, A.; Rai, T.; Sasaki, S.; Uchida, S. Molecular mechanisms of Bartter syndrome caused by mutations in the BSND gene. Histochem. Cell Biol. 2003, 119, 485–493. [Google Scholar] [CrossRef]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef]
- Jeck, N.; Waldegger, P.; Doroszewicz, J.; Seyberth, H.; Waldegger, S. A common sequence variation of the CLCNKB gene strongly activates ClC-Kb chloride channel activity. Kidney Int. 2004, 65, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.; Lampert, A.; Waldegger, S.; Jeck, N.; Waldegger, P.; Artunc, F.; Seebohm, G.; Lang, U.E.; Kupka, S.; Pfister, M.; et al. Influence of gain of function epithelial chloride channel ClC-Kb mutation on hearing thresholds. Hear. Res. 2006, 214, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Barlassina, C.; Fiume, C.D.; Lanzani, C.; Manunta, P.; Guffanti, G.; Ruello, A.; Bianchi, G.; Del Vecchio, L.; Macciardi, F.; Cusi, D. Common genetic variants and haplotypes in renal CLCNKA gene are associated to salt-sensitive hypertension. Hum. Mol. Genet. 2007, 16, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Joo, K.; Oh, J.; Han, J.H.; Park, H.-R.; Lee, S.; Oh, D.-Y.; Woo, S.J.; Choi, B.Y. Severe or Profound Sensorineural Hearing Loss Caused by Novel USH2A Variants in Korea: Potential Genotype-Phenotype Correlation. Clin. Exp. Otorhinolaryngol. 2020, 13, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Kim, M.Y.; Han, J.H.; Park, S.S.; Yun, Y.; Jee, S.-C.; Han, J.J.; Lee, J.H.; Seok, H.; Choi, B.Y. Ramifications of POU4F3 variants associated with autosomal dominant hearing loss in various molecular aspects. Sci. Rep. 2023, 13, 1–14. [Google Scholar] [CrossRef]
- Lee, S.; Yun, Y.; Cha, J.H.; Han, J.H.; Lee, D.H.; Song, J.-J.; Park, M.K.; Lee, J.H.; Oh, S.H.; Choi, B.Y.; et al. Phenotypic and molecular basis of SIX1 variants linked to non-syndromic deafness and atypical branchio-otic syndrome in South Korea. Sci. Rep. 2023, 13, 11776. [Google Scholar] [CrossRef]
- Jo, H.D.; Han, J.H.; Lee, S.M.; Choi, D.H.; Lee, S.-Y.; Choi, B.Y. Genetic Load of Alternations of Transcription Factor Genes in Non-Syndromic Deafness and the Associated Clinical Phenotypes: Experience from Two Tertiary Referral Centers. Biomedicines 2022, 10, 2125. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Gene | Variant | Zygosity | Age | Clinical Phenotypes | ||
|---|---|---|---|---|---|---|---|
| Robitaille et al., 2011 [20] | CLCNKB | c.229G>C:p. Ala77Pro | Homozygote | 13-year-old | Batter syndrome type III | Renal phenotype | Severe dehydration secondary to a prolonged diarrhea Chronic metabolic alkalosis Normal renal function |
| Hearing phenotype | Sensorineural deafness with a loss of 60–70 dB from 250 to 9 kHz in both ears | ||||||
| Nozu et al., 2008 [12] | CLCNKA | c.778C>T:p.Gln260Ter | Heterozygote | 2-year-old | Batter syndrome type IV | Renal phenotype | Polyuria and severe volume depletion Hyponatremia/normal potassium concentration Severe hypokalemia and metabolic alkalosis Acute renal failure. |
| Deletion (CLCNKA exon16- CLCNKB intron2) | Heterozygote | ||||||
| CLCNKB | IVS17+1G>A: Splicing variant | Heterozygote | Hearing phenotype | Bilateral sensorineural deafness | |||
| Deletion (CLCNKA exon16- CLCNKB intron2) | Heterozygote | ||||||
| Schlingmann et al., 2004 [9] | CLCNKA | c.240G>C:p.Trp80Cys | Homozygote | 2-month-old | Batter syndrome type IV | Renal phenotype | Polyuria and volume depletion associated with hypokalemia and metabolic alkalosis |
| CLCNKB | Whole gene deletion | Homozygote | Hearing phenotype | Bilateral sensorineural deafness | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, Y.; Park, S.S.; Lee, S.; Seok, H.; Park, S.; Lee, S.-Y. Expanding Genotype–Phenotype Correlation of CLCNKA and CLCNKB Variants Linked to Hearing Loss. Int. J. Mol. Sci. 2023, 24, 17077. https://doi.org/10.3390/ijms242317077
Yun Y, Park SS, Lee S, Seok H, Park S, Lee S-Y. Expanding Genotype–Phenotype Correlation of CLCNKA and CLCNKB Variants Linked to Hearing Loss. International Journal of Molecular Sciences. 2023; 24(23):17077. https://doi.org/10.3390/ijms242317077
Chicago/Turabian StyleYun, Yejin, Sang Soo Park, Soyoung Lee, Heeyoung Seok, Seongyeol Park, and Sang-Yeon Lee. 2023. "Expanding Genotype–Phenotype Correlation of CLCNKA and CLCNKB Variants Linked to Hearing Loss" International Journal of Molecular Sciences 24, no. 23: 17077. https://doi.org/10.3390/ijms242317077