
The Nexus of Inflammation-Induced Epithelial-Mesenchymal Transition and Lung Cancer Progression: A Roadmap to Pentacyclic Triterpenoid-Based Therapies

Abstract
:1. Introduction
2. Inflammatory-Driven EMT of Lung Cancer Cells: Key Players and Regulators
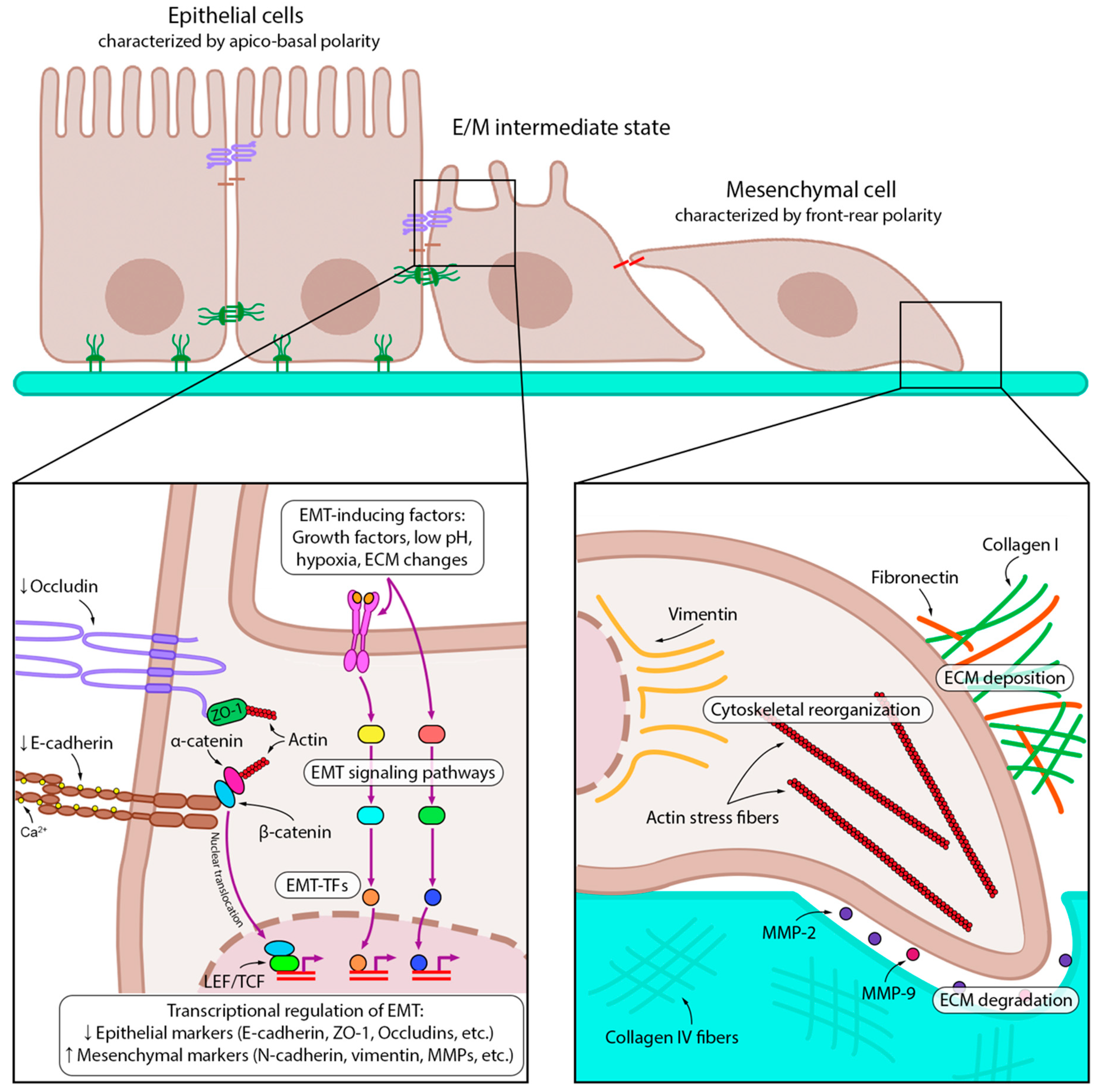
2.1. EMT-Associated Changes in Cancer Cells
2.2. Inflammatory TME and Its Role in EMT Induction in Lung Cancer Cells
2.2.1. Cancer-Associated Fibroblasts
2.2.2. Immune Cells
Granulocytes
Tumor-Associated Macrophages
Mast Cells
2.2.3. Other Pro-Inflammatory EMT-Inducing Mediators in iTME
2.2.4. ECM Components as EMT Regulators
2.2.5. Reactive Oxygen Species in the Regulation of EMT in Lung Cells
2.2.6. COVID-19-Associated Inducers of EMT in Lung Tissue
3. Inflammation-Induced EMT as a Source of Cancer Stem Cells
4. Clinical Trials of Drugs Targeting Key Regulators of Inflammation-Driven EMT
5. Inhibitory Effect of Triterpenoids on Inflammation-Induced EMT in Lung Cancer Cells
5.1. Ursane-Type Triterpenoids
5.2. Oleanane- and Friedelane-Type Triterpenoids
5.3. Lupane-Type Triterpenoids
6. Inhibitory Effect of Triterpenoids on Stem-like Properties of Lung Cancer Cells
7. Future Prospective and Limitations
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Arseniev, A.I.; Nefedov, A.O.; Novikov, S.N.; Barchuk, A.A.; Tarkov, S.A.; Kostitsin, K.A.; Nefedova, A.V.; Aristidov, N.Y.; Semiletova, Y.V.; Ryazankina, A.A. Algorithms of non-invasive, minimally-invasive and invasive diagnostics for lung cancer (review). Prev. Clin. Med. 2021, 2, 69–77. (In Russian) [Google Scholar]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Menju, T.; Date, H. Lung cancer and epithelial-mesenchymal transition. Gen. Thorac. Cardiovasc. Surg. 2021, 69, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Tsoukalas, N.; Aravantinou-Fatorou, E.; Tolia, M.; Giaginis, C.; Galanopoulos, M.; Kiakou, M.; Kostakis, I.D.; Dana, E.; Vamvakaris, I.; Korogiannos, A.; et al. Epithelial-Mesenchymal Transition in Non Small-cell Lung Cancer. Anticancer Res. 2017, 37, 1773–1778. [Google Scholar] [CrossRef] [PubMed]
- Trandafir, L.M.; Miron, O.; Afrasanie, V.-A.; Paduraru, M.-I.; Miron, L. The relationship between chronic lung diseases and lung cancer-a narrative review. JBUON 2020, 25, 1687–1692. [Google Scholar]
- Szalontai, K.; Gémes, N.; Furák, J.; Varga, T.; Neuperger, P.; Balog, J.Á.; Puskás, L.G.; Szebeni, G.J. Chronic Obstructive Pulmonary Disease: Epidemiology, Biomarkers, and Paving the Way to Lung Cancer. J. Clin. Med. 2021, 10, 2889. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, M.Q.; Walters, E.H.; Shukla, S.D.; Weston, S.; Muller, H.K.; Ward, C.; Sohal, S.S. β-catenin, Twist and Snail: Transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Pandolfi, L.; Bozzini, S.; Frangipane, V.; Percivalle, E.; De Luigi, A.; Violatto, M.B.; Lopez, G.; Gabanti, E.; Carsana, L.; D’Amato, M.; et al. Neutrophil Extracellular Traps Induce the Epithelial-Mesenchymal Transition: Implications in Post-COVID-19 Fibrosis. Front. Immunol. 2021, 12, 663303. [Google Scholar] [CrossRef]
- Markov, A.V.; Zenkova, M.A.; Logashenko, E.B. Modulation of Tumour-Related Signaling Pathways by Natural Pentacyclic Triterpenoids and their Semisynthetic Derivatives. Curr. Med. Chem. 2017, 24, 1277–1320. [Google Scholar] [CrossRef]
- Kozak, J.; Forma, A.; Czeczelewski, M.; Kozyra, P.; Sitarz, E.; Radzikowska-Büchner, E.; Sitarz, M.; Baj, J. Inhibition or Reversal of the Epithelial-Mesenchymal Transition in Gastric Cancer: Pharmacological Approaches. Int. J. Mol. Sci. 2021, 22, 277. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.L.; Montes de Oca, M.K.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial–mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Y.; Li, Y.; Tang, Y.T.; Ma, X.D.; Tang, Z.Y. Anticancer activity of oleanolic acid and its derivatives: Recent advances in evidence, target profiling and mechanisms of action. Biomed. Pharmacother. 2022, 145, 112397. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Q.; Zhou, C.C.; Yu, L.Y.; Wang, L.; Deng, J.L.; Tao, Y.L.; Zhang, F.; Chen, W.S. Natural product derived phytochemicals in managing acute lung injury by multiple mechanisms. Pharmacol. Res. 2021, 163, 105224. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Malzone, M.G.; Vanacore, D.; Di Franco, R.; La Mantia, E.; Iovane, G.; Piscitelli, R.; et al. Epithelial-mesenchymal transition in prostate cancer: An overview. Oncotarget 2017, 8, 35376–35389. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, Y.L.; Liang, X.H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar] [CrossRef]
- Suzuki, A.; Maeda, T.; Baba, Y.; Shimamura, K.; Kato, Y. Acidic extracellular ph promotes epithelial mesenchymal transition in lewis lung carcinoma model. Cancer Cell Int. 2014, 14, 129. [Google Scholar] [CrossRef]
- Deng, Z.; Fear, M.W.; Suk Choi, Y.; Wood, F.M.; Allahham, A.; Mutsaers, S.E.; Prêle, C.M. The extracellular matrix and mechanotransduction in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2020, 126, 105802. [Google Scholar] [CrossRef]
- Debnath, P.; Huirem, R.S.; Dutta, P.; Palchaudhuri, S. Epithelial–mesenchymal transition and its transcription factors. Biosci. Rep. 2021, 42, BSR20211754. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.Y.; Lin, H.-H.H.; Tang, M.-J.J.; Wang, Y.-K.K. Vimentin contributes to epithelial-mesenchymal transition ancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Griggs, L.A.; Hassan, N.T.; Malik, R.S.; Griffin, B.P.; Martinez, B.A.; Elmore, L.W.; Lemmon, C.A. Fibronectin fibrils regulate TGF-β1-induced Epithelial-Mesenchymal Transition. Matrix Biol. 2017, 60–61, 157–175. [Google Scholar] [CrossRef]
- Zhang, K.; Corsa, C.A.; Ponik, S.M.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.W.; Keely, P.J.; Longmore, G.D. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Rajasegaran, T.; How, C.W.; Saud, A.; Ali, A.; Lim, J.C. Targeting Inflammation in Non-Small Cell Lung Cancer through Drug Repurposing. Pharmaceuticals 2023, 16, 451. [Google Scholar] [CrossRef]
- Houghton, A.M.G. Mechanistic links between COPD and lung cancer. Nat. Rev. Cancer 2013, 13, 233–245. [Google Scholar] [CrossRef]
- Samet, J.M. Does idiopathic pulmonary fibrosis increase lung cancer risk? Am. J. Respir. Crit. Care Med. 2000, 161, 1–2. [Google Scholar] [CrossRef]
- Ahmad, S.; Manzoor, S.; Siddiqui, S.; Mariappan, N.; Zafar, I.; Ahmad, A.; Ahmad, A. Epigenetic underpinnings of inflammation: Connecting the dots between pulmonary diseases, lung cancer and COVID-19. Semin. Cancer Biol. 2022, 83, 384–398. [Google Scholar] [CrossRef]
- Islam, M.S.; Morshed, M.R.; Babu, G.; Khan, M.A. The role of inflammations and EMT in carcinogenesis. Adv. Cancer Biol.-Metastasis 2022, 5, 100055. [Google Scholar] [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, E.L.; Walser, T.C.; Krysan, K.; Liclican, E.L.; Grant, J.L.; Rodriguez, N.L.; Dubinett, S.M. The Inflammatory Tumor Microenvironment, Epithelial Mesenchymal Transition and Lung Carcinogenesis. Cancer Microenviron. 2012, 5, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Abulaiti, A.; Kimura, T.; Funaki, S.; Nakagiri, T.; Inoue, M.; Sawabata, N.; Minami, M.; Morii, E.; Okumura, M. Pulmonary Fibroblasts Induce Epithelial Mesenchymal Transition and Some Characteristics of Stem Cells in Non-Small Cell Lung Cancer. Ann. Thorac. Surg. 2013, 96, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zeng, S.; Wang, Z.; Wu, M.; Ma, Y.; Ye, X.; Zhang, B.; Liu, H. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef]
- Wang, L.; Cao, L.; Wang, H.; Liu, B.; Zhang, Q.; Meng, Z.; Wu, X.; Zhou, Q.; Xu, K. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget 2017, 8, 76116–76128. [Google Scholar] [CrossRef]
- Wang, Y.; Lan, W.; Xu, M.; Song, J.; Mao, J.; Li, C.; Du, X.; Jiang, Y.; Li, E.; Zhang, R.; et al. Cancer-associated fibroblast-derived SDF-1 induces epithelial-mesenchymal transition of lung adenocarcinoma via CXCR4/β-catenin/PPARδ signalling. Cell Death Dis. 2021, 12, 214. [Google Scholar] [CrossRef]
- Ren, Y.; Cao, L.; Wang, L.; Zheng, S.; Zhang, Q.; Guo, X.; Li, X.; Chen, M.; Wu, X.; Furlong, F.; et al. Autophagic secretion of HMGB1 from cancer-associated fibroblasts promotes metastatic potential of non-small cell lung cancer cells via NFκB signaling. Cell Death Dis. 2021, 12, 858. [Google Scholar] [CrossRef]
- You, J.; Li, M.; Cao, L.M.; Gu, Q.H.; Deng, P.B.; Tan, Y.; Hu, C.P. Snail1-dependent cancer-associated fibroblasts induce epithelial-mesenchymal transition in lung cancer cells via exosomes. QJM Int. J. Med. 2019, 112, 581–590. [Google Scholar] [CrossRef]
- Yang, F.; Yan, Y.; Yang, Y.; Hong, X.; Wang, M.; Yang, Z.; Liu, B.; Ye, L. MiR-210 in exosomes derived from CAFs promotes non-small cell lung cancer migration and invasion through PTEN/PI3K/AKT pathway. Cell. Signal. 2020, 73, 109675. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Ambati, R.; Gundamaraju, R. Exploring the Crosstalk between Inflammation and Epithelial-Mesenchymal Transition in Cancer. Mediat. Inflamm. 2021, 2021, 9918379. [Google Scholar] [CrossRef]
- Hui, X.; Al-Ward, H.; Shaher, F.; Liu, C.-Y.; Liu, N. The Role of miR-210 in the Biological System: A Current Overview. Hum. Hered. 2020, 84, 233–239. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune- modulatory properties in cancer cells. Br. J. Cancer 2015, 116, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Worthen, G.S.; Albelda, S.M. Polarization of TAN phenotype by TGFb: “N1” versus “N2” TAN. Cancer Cell 2010, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Shen, M.; Zhang, P.; Zheng, C.; Pang, Z.; Zhu, L.; Du, J. Intratumoral neutrophil granulocytes contribute to epithelial-mesenchymal transition in lung adenocarcinoma cells. Tumor Biol. 2015, 36, 7789–7796. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, A.; Hosoki, K.; Toda, M.; Miyake, Y.; Matsushima, Y.; Matsumoto, T.; Boveda-Ruiz, D.; Gil-Bernabe, P.; Nagao, M.; Sugimoto, M.; et al. Eosinophils Promote Epithelial to Mesenchymal Transition of Bronchial Epithelial Cells. PLoS ONE 2013, 8, e64281. [Google Scholar] [CrossRef]
- Hosoki, K.; Kainuma, K.; Toda, M.; Harada, E.; Chelakkot-Govindalayathila, A.L.; Roeen, Z.; Nagao, M.; D’Alessandro-Gabazza, C.N.; Fujisawa, T.; Gabazza, E.C. Montelukast suppresses epithelial to mesenchymal transition of bronchial epithelial cells induced by eosinophils. Biochem. Biophys. Res. Commun. 2014, 449, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Trinh, H.K.T.; Lee, S.-H.; Cao, T.B.T.; Park, H.-S. Asthma pharmacotherapy: An update on leukotriene treatments. Expert Rev. Respir. Med. 2019, 13, 1169–1178. [Google Scholar] [CrossRef]
- Kim, S.Y.; Nair, M.G. Macrophages in wound healing: Activation and plasticity. Immunol. Cell Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gao, Q.; Han, S.; Pan, F.; Fan, W. The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-Twist signaling. Tumor Biol. 2014, 36, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Dehai, C.; Bo, P.; Qiang, T.; Lihua, S.; Fang, L.; Shi, J.; Jingyan, C.; Yan, Y.; Guangbin, W.; Zhenjun, Y. Enhanced invasion of lung adenocarcinoma cells after co-culture with THP-1-derived macrophages via the induction of EMT by IL-6. Immunol. Lett. 2014, 160, 1–10. [Google Scholar] [CrossRef]
- Qin, Y.; Zhao, P.; Chen, Y.; Liu, X.; Dong, H.; Zheng, W.; Li, C.; Mao, X.; Li, J. Lipopolysaccharide induces epithelial–mesenchymal transition of alveolar epithelial cells cocultured with macrophages possibly via the JAK2/STAT3 signaling pathway. Hum. Exp. Toxicol. 2020, 39, 224–234. [Google Scholar] [CrossRef]
- Shang, G.-S.; Liu, L.; Qin, Y.-W. IL-6 and TNF-α promote metastasis of lung cancer by inducing epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 4657–4660. [Google Scholar] [CrossRef]
- Yin, Y.; Shelke, G.V.; Lässer, C.; Brismar, H.; Lötvall, J. Extracellular vesicles from mast cells induce mesenchymal transition in airway epithelial cells. Respir. Res. 2020, 21, 101. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, Y.; Hardie, W.J.; Zhou, X. Mast cell chymase affects the proliferation and metastasis of lung carcinoma cells in vitro. Oncol. Lett. 2017, 14, 3193–3198. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ong, S.L.; Tran, L.M.; Jing, Z.; Liu, B.; Park, S.J.; Huang, Z.L.; Walser, T.C.; Heinrich, E.L.; Lee, G.; et al. Chronic IL-1 β -induced inflammation regulates epithelial- to-mesenchymal transition memory phenotypes via epigenetic modifications in non-small cell lung cancer. Sci. Rep. 2020, 10, 377. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Han, J.; Fan, J.; Duan, L.; Guo, M.; Lv, Z.; Hu, G.; Chen, L.; Wu, F.; Tao, X.; et al. IL-17 induces EMT via Stat3 in lung adenocarcinoma. Am. J. Cancer Res. 2016, 6, 440–451. [Google Scholar] [PubMed]
- Liu, X. Inflammatory cytokines augments TGF-β1-induced epithelial-mesenchymal transition in A549 cells by up-regulating TβR-I. Cell Motil. Cytoskelet. 2008, 65, 935–944. [Google Scholar] [CrossRef]
- Li, Q.; Han, Y.; Fei, G.; Guo, Z.; Ren, T.; Liu, Z. IL-17 promoted metastasis of non-small-cell lung cancer cells. Immunol. Lett. 2012, 148, 144–150. [Google Scholar] [CrossRef]
- Gu, K.; Li, M.-M.; Shen, J.; Liu, F.; Cao, J.-Y.; Jin, S.; Yu, Y. Interleukin-17-induced EMT promotes lung cancer cell migration and invasion via NF-κB/ZEB1 signal pathway. Am. J. Cancer Res. 2015, 5, 1169–1179. [Google Scholar] [PubMed]
- Kumar, M.; Allison, D.F.; Baranova, N.N.; Wamsley, J.J.; Katz, A.J.; Bekiranov, S.; Jones, D.R.; Mayo, M.W. NF-κB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS ONE 2013, 8, e68597. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Yatabe, Y.; Achiwa, H.; Muramatsu, H.; Kozaki, K.I.; Nakamura, S.; Ogawa, M.; Mitsudomi, T.; Sugiura, T.; Takahashi, T. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res. 1998, 58, 3761–3764. [Google Scholar]
- Dohadwala, M.; Yang, S.C.; Luo, J.; Sharma, S.; Batra, R.K.; Huang, M.; Lin, Y.; Goodglick, L.; Krysan, K.; Fishbein, M.C.; et al. Cyclooxygenase-2-dependent regulation of E-cadherin: Prostaglandin E 2 induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res. 2006, 66, 5338–5345. [Google Scholar] [CrossRef]
- Takai, E.; Tsukimoto, M.; Kojima, S. TGF-β1 Downregulates COX-2 Expression Leading to Decrease of PGE2 Production in Human Lung Cancer A549 Cells, Which Is Involved in Fibrotic Response to TGF-β1. PLoS ONE 2013, 8, e76346. [Google Scholar] [CrossRef]
- Vafaeinik, F.; Kum, H.J.; Jin, S.Y.; Min, D.S.; Song, S.H.; Ha, H.K.; Kim, C.D.; Bae, S.S. Regulation of Epithelial-Mesenchymal Transition of A549 Cells by Prostaglandin D2. Cell. Physiol. Biochem. 2022, 56, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Petrey, A.; de la Motte, C. Hyaluronan, a Crucial Regulator of Inflammation. Front. Immunol. 2014, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Marozzi, M.; Parnigoni, A.; Negri, A.; Viola, M.; Vigetti, D.; Passi, A.; Karousou, E.; Rizzi, F. Inflammation, Extracellular Matrix Remodeling, and Proteostasis in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 8102. [Google Scholar] [CrossRef]
- Chow, G.; Tauler, J.; Mulshine, J.L. Cytokines and Growth Factors Stimulate Hyaluronan Production: Role of Hyaluronan in Epithelial to Mesenchymal-Like Transition in Non-Small Cell Lung Cancer. J. Biomed. Biotechnol. 2010, 2010, 485468. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-J.; Hsu, S. Acquisition of epithelial–mesenchymal transition and cancer stem-like phenotypes within chitosan-hyaluronan membrane-derived 3D tumor spheroids. Biomaterials 2014, 35, 10070–10079. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-W.; Hsu, S. Chitosan-hyaluronan based 3D co-culture platform for studying the crosstalk of lung cancer cells and mesenchymal stem cells. Acta Biomater. 2016, 42, 157–167. [Google Scholar] [CrossRef]
- Suda, K.; Murakami, I.; Yu, H.; Kim, J.; Tan, A.-C.; Mizuuchi, H.; Rozeboom, L.; Ellison, K.; Rivard, C.J.; Mitsudomi, T.; et al. CD44 Facilitates Epithelial-to-Mesenchymal Transition Phenotypic Change at Acquisition of Resistance to EGFR Kinase Inhibitors in Lung Cancer. Mol. Cancer Ther. 2018, 17, 2257–2265. [Google Scholar] [CrossRef]
- Chen, F.; Zhu, X.; Zheng, J.; Xu, T.; Wu, K.; Ru, C. RHAMM regulates the growth and migration of lung adenocarcinoma A549 cell line by regulating Cdc2/CyclinB1 and MMP9 genes. Math. Biosci. Eng. 2020, 17, 2150–2163. [Google Scholar] [CrossRef]
- Su, J.; Wu, S.; Wu, H.; Li, L.; Guo, T. CD44 is functionally crucial for driving lung cancer stem cells metastasis through Wnt/β-catenin-FoxM1-Twist signaling. Mol. Carcinog. 2016, 55, 1962–1973. [Google Scholar] [CrossRef]
- Li, L.; Qi, L.; Liang, Z.; Song, W.; Liu, Y.; Wang, Y.; Sun, B.; Zhang, B.; Cao, W. Transforming growth factor-β1 induces EMT by the transactivation of epidermal growth factor signaling through HA/CD44 in lung and breast cancer cells. Int. J. Mol. Med. 2015, 36, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Nurwidya, F.; Takahashi, F.; Kato, M.; Baskoro, H.; Hidayat, M.; Wirawan, A.; Takahashi, K. CD44 silencing decreases the expression of stem cell-related factors induced by transforming growth factor β1 and tumor necrosis factor α in lung cancer: Preliminary findings. Bosn. J. Basic Med. Sci. 2017, 17, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Huang, C.-S.; Yang, Y.-P.; Liu, C.-Y.; Liu, Y.-Y.; Wu, W.-W.; Lu, K.-H.; Chen, K.-H.; Chang, Y.-L.; Lee, S.-D.; et al. The subpopulation of CD44-positive cells promoted tumorigenicity and metastatic ability in lung adenocarcinoma. J. Chin. Med. Assoc. 2019, 82, 196–201. [Google Scholar] [CrossRef]
- Monleón-Guinot, I.; Milian, L.; Martínez-Vallejo, P.; Sancho-Tello, M.; Llop-Miguel, M.; Galbis, J.M.; Cremades, A.; Carda, C.; Mata, M. Morphological Characterization of Human Lung Cancer Organoids Cultured in Type I Collagen Hydrogels: A Histological Approach. Int. J. Mol. Sci. 2023, 24, 10131. [Google Scholar] [CrossRef]
- Lotsberg, M.L.; Røsland, G.V.; Rayford, A.J.; Dyrstad, S.E.; Ekanger, C.T.; Lu, N.; Frantz, K.; Stuhr, L.E.B.; Ditzel, H.J.; Thiery, J.P.; et al. Intrinsic Differences in Spatiotemporal Organization and Stromal Cell Interactions Between Isogenic Lung Cancer Cells of Epithelial and Mesenchymal Phenotypes Revealed by High-Dimensional Single-Cell Analysis of Heterotypic 3D Spheroid Models. Front. Oncol. 2022, 12, 818437. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.H.; Ungewiss, C.; Tong, P.; Byers, L.A.; Wang, J.; Canales, J.R.; Villalobos, P.A.; Uraoka, N.; Mino, B.; Behrens, C.; et al. ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene 2017, 36, 1925–1938. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Maeda, M.; Chaika, N.; Johnson, K.R.; Wheelock, M.J. Collagen I Promotes Epithelial-to-Mesenchymal Transition in Lung Cancer Cells via Transforming Growth Factor–β Signaling. Am. J. Respir. Cell Mol. Biol. 2008, 38, 95–104. [Google Scholar] [CrossRef]
- Liu, C.-C.; Lin, J.-H.; Hsu, T.-W.; Hsu, J.-W.; Chang, J.-W.; Su, K.; Hsu, H.-S.; Hung, S.-C. Collagen XVII/laminin-5 activates epithelial-to-mesenchymal transition and is associated with poor prognosis in lung cancer. Oncotarget 2018, 9, 1656–1672. [Google Scholar] [CrossRef]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.-H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef]
- Liu, L.; Stephens, B.; Bergman, M.; May, A.; Chiang, T. Role of Collagen in Airway Mechanics. Bioengineering 2021, 8, 13. [Google Scholar] [CrossRef]
- Jones, V.A.; Patel, P.M.; Gibson, F.T.; Cordova, A.; Amber, K.T. The Role of Collagen XVII in Cancer: Squamous Cell Carcinoma and Beyond. Front. Oncol. 2020, 10, 353. [Google Scholar] [CrossRef]
- Hasan Ali, O.; Bomze, D.; Ring, S.S.; Berner, F.; Fässler, M.; Diem, S.; Abdou, M.-T.; Hammers, C.; Emtenani, S.; Braun, A.; et al. BP180-specific IgG is associated with skin adverse events, therapy response, and overall survival in non-small cell lung cancer patients treated with checkpoint inhibitors. J. Am. Acad. Dermatol. 2020, 82, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Hu, S.; Tang, C.; Liu, G. Mesenchymal stem cells promote cell invasion and migration and autophagy-induced epithelial-mesenchymal transition in A549 lung adenocarcinoma cells. Cell Biochem. Funct. 2018, 36, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Shi, F.; Zhai, R.; Wang, H.; Li, K.; Xu, C.; Yao, W.; Zhou, F. TGF-β promote epithelial-mesenchymal transition via NF-κB/NOX4/ROS signal pathway in lung cancer cells. Mol. Biol. Rep. 2021, 48, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Lee, J.-H.; Kwak, M.-K. NRF2 level is negatively correlated with TGF-β1-induced lung cancer motility and migration via NOX4-ROS signaling. Arch. Pharm. Res. 2020, 43, 1297–1310. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; He, K.; Wang, D.; Yuan, X.; Liu, Y.; Ji, H.; Song, J. TMEPAI regulates EMT in lung cancer cells by modulating the ROS and IRS-1 signaling pathways. Carcinogenesis 2013, 34, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Xiang, Z.; Wu, Y.; Gu, Y.; Guo, J.; Geng, F. Analysis of the susceptibility of lung cancer patients to SARS-CoV-2 infection. Mol. Cancer 2020, 19, 80. [Google Scholar] [CrossRef]
- Zhang, H.; Quek, K.; Chen, R.; Chen, J.; Chen, B. Expression of the SAR2-Cov-2 receptor ACE2 reveals the susceptibility of COVID-19 in non-small cell lung cancer. J. Cancer 2020, 11, 5289–5292. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Samarelli, A.V.; Tonelli, R.; Cerri, S.; Clini, E.; Stella, F.; Dominici, M. Biological effects of COVID-19 on lung cancer: Can we drive our decisions. Front. Oncol. 2022, 12, 1029830. [Google Scholar] [CrossRef]
- Saygideger, Y.; Sezan, A.; Candevir, A.; Saygıdeğer Demir, B.; Güzel, E.; Baydar, O.; Derinoz, E.; Komur, S.; Kuscu, F.; Ozyılmaz, E.; et al. COVID-19 patients’ sera induce epithelial mesenchymal transition in cancer cells. Cancer Treat. Res. Commun. 2021, 28, 100406. [Google Scholar] [CrossRef]
- Falleni, M.; Tosi, D.; Savi, F.; Chiumello, D.; Bulfamante, G. Endothelial-Mesenchymal Transition in COVID-19 lung lesions. Pathol.-Res. Pract. 2021, 221, 153419. [Google Scholar] [CrossRef]
- Jahankhani, K.; Ahangari, F.; Adcock, I.M.; Mortaz, E. Possible cancer-causing capacity of COVID-19: Is SARS-CoV-2 an oncogenic agent? Biochimie 2023, 213, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.; Deng, X.; Wang, P.; Mo, Y.; Zhang, Y.; Tong, X. Cytokines as drivers: Unraveling the mechanisms of epithelial-mesenchymal transition in COVID-19 lung fibrosis. Biochem. Biophys. Res. Commun. 2023, 686, 149118. [Google Scholar] [CrossRef] [PubMed]
- Pi, P.; Zeng, Z.; Zeng, L.; Han, B.; Bai, X.; Xu, S. Molecular mechanisms of COVID-19-induced pulmonary fibrosis and epithelial-mesenchymal transition. Front. Pharmacol. 2023, 14, 1218059. [Google Scholar] [CrossRef] [PubMed]
- Liapis, I.; Baritaki, S. COVID-19 vs. Cancer Immunosurveillance: A Game of Thrones within an Inflamed Microenviroment. Cancers 2022, 14, 4330. [Google Scholar] [CrossRef]
- du Plessis, M.; Fourie, C.; Riedemann, J.; de Villiers, W.J.S.; Engelbrecht, A.M. Cancer and Covid-19: Collectively catastrophic. Cytokine Growth Factor Rev. 2022, 63, 78–89. [Google Scholar] [CrossRef]
- Leon, G.; MacDonagh, L.; Finn, S.P.; Cuffe, S.; Barr, M.P. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signaling pathways. Pharmacol. Ther. 2016, 158, 71–90. [Google Scholar] [CrossRef]
- Heng, W.S.; Gosens, R.; Kruyt, F.A.E. Lung cancer stem cells: Origin, features, maintenance mechanisms and therapeutic targeting. Biochem. Pharmacol. 2019, 160, 121–133. [Google Scholar] [CrossRef]
- Zhang, F.; Li, T.; Han, L.; Qin, P.; Wu, Z.; Xu, B.; Gao, Q.; Song, Y. TGFβ1-induced down-regulation of microRNA-138 contributes to epithelial-mesenchymal transition in primary lung cancer cells. Biochem. Biophys. Res. Commun. 2018, 496, 1169–1175. [Google Scholar] [CrossRef]
- Kahm, Y.-J.; Kim, R.-K.; Jung, U.; Kim, I.-G. Epithelial membrane protein 3 regulates lung cancer stem cells via the TGF-β signaling pathway. Int. J. Oncol. 2021, 59, 80. [Google Scholar] [CrossRef]
- Yang, L.; Dong, Y.; Li, Y.; Wang, D.; Liu, S.; Wang, D.; Gao, Q.; Ji, S.; Chen, X.; Lei, Q.; et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1 pathway in non-small cell lung cancer. Int. J. Cancer 2019, 145, 1099–1110. [Google Scholar] [CrossRef]
- Wamsley, J.J.; Kumar, M.; Allison, D.F.; Clift, S.H.; Holzknecht, C.M.; Szymura, S.J.; Hoang, S.A.; Xu, X.; Moskaluk, C.A.; Jones, D.R.; et al. Activin upregulation by NF-κB is required to maintain mesenchymal features of cancer stem-like cells in non-small cell lung cancer. Cancer Res. 2015, 75, 426–435. [Google Scholar] [CrossRef]
- Song, M.; Ping, Y.; Zhang, K.; Yang, L.; Li, F.; Zhang, C.; Cheng, S.; Yue, D.; Maimela, N.R.; Qu, J.; et al. Low-Dose IFNγ Induces Tumor Cell Stemness in Tumor Microenvironment of Non–Small Cell Lung Cancer. Cancer Res. 2019, 79, 3737–3748. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Yang, X.; Duan, S.; Tsai, Y.; Strojny, L.R.; Keng, P.; Chen, Y. IL-6 promotes growth and epithelial-mesenchymal transition of CD133+ cells of non-small cell lung cancer. Oncotarget 2016, 7, 6626–6638. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Lin, J.-H.; Hsu, T.-W.; Su, K.; Li, A.F.-Y.; Hsu, H.-S.; Hung, S.-C. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int. J. Cancer 2015, 136, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yang, L.; Zhang, C.; Wang, R.; Zhang, Z.; He, Q.; Chen, X.; Zhang, B.; Qin, Z.; Wang, L.; et al. Th17 cell-derived IL-17A promoted tumor progression via STAT3/NF-κB/Notch1 signaling in non-small cell lung cancer. Oncoimmunology 2018, 7, e1461303. [Google Scholar] [CrossRef]
- Lu, C.-H.; Yeh, D.-W.; Lai, C.-Y.; Liu, Y.-L.; Huang, L.-R.; Lee, A.Y.-L.; Jin, S.-L.C.; Chuang, T.-H. USP17 mediates macrophage-promoted inflammation and stemness in lung cancer cells by regulating TRAF2/TRAF3 complex formation. Oncogene 2018, 37, 6327–6340. [Google Scholar] [CrossRef]
- Zakaria, N.; Mohd Yusoff, N.; Zakaria, Z.; Widera, D.; Yahaya, B.H. Inhibition of NF-κB signaling reduces the stemness characteristics of lung cancer stem cells. Front. Oncol. 2018, 8, 166. [Google Scholar] [CrossRef]
- Zeni, E.; Mazzetti, L.; Miotto, D.; Lo Cascio, N.; Maestrelli, P.; Querzoli, P.; Pedriali, M.; De Rosa, E.; Fabbri, L.M.; Mapp, C.E.; et al. Macrophage expression of interleukin-10 is a prognostic factor in nonsmall cell lung cancer. Eur. Respir. J. 2007, 30, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Tang, S.-J.; Sun, G.-H.; Sun, K.-H. CXCR7 mediates TGFβ1-promoted EMT and tumor-initiating features in lung cancer. Oncogene 2016, 35, 2123–2132. [Google Scholar] [CrossRef]
- Chang, Y.-W.; Su, Y.-J.; Hsiao, M.; Wei, K.-C.; Lin, W.-H.; Liang, C.-J.; Chen, S.-C.; Lee, J.-L. Diverse targets of β-catenin during the epithelial–mesenchymal transition define cancer stem cells and predict disease relapse. Cancer Res. 2015, 75, 3398–3410. [Google Scholar] [CrossRef]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S.; Hwang, L.; Trieu, V.N. Recurrent or Refractory High-Grade Gliomas Treated by Convection-Enhanced Delivery of a TGFβ2-Targeting RNA Therapeutic: A Post-Hoc Analysis with Long-Term Follow-Up. Cancers 2019, 11, 1891. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Bazhenova, L.A.; Nemunaitis, J.; Tan, M.; Juhász, E.; Ramlau, R.; van den Heuvel, M.M.; Lal, R.; Kloecker, G.H.; Eaton, K.D.; et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro. Oncol. 2016, 18, 1146–1156. [Google Scholar] [CrossRef]
- Giannelli, G.; Santoro, A.; Kelley, R.K.; Gane, E.; Paradis, V.; Cleverly, A.; Smith, C.; Estrem, S.T.; Man, M.; Wang, S.; et al. Biomarkers and overall survival in patients with advanced hepatocellular carcinoma treated with TGF-βRI inhibitor galunisertib. PLoS ONE 2020, 15, e0222259. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Chung, C.-L.; Hu, T.-H.; Chen, J.-J.; Liu, P.-F.; Chen, C.-L. Recent progress in TGF-β inhibitors for cancer therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFβ Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, X.; Lu, L.; Yuan, H.; Wang, Y.; Chen, Z.; Ji, R.; Zhou, Y. Comprehensive evaluation of clinical efficacy and safety of celecoxib combined with chemotherapy in management of gastric cancer. Medicine 2017, 96, e8857. [Google Scholar] [CrossRef]
- Legge, F.; Paglia, A.; D’Asta, M.; Fuoco, G.; Scambia, G.; Ferrandina, G. Phase II study of the combination carboplatin plus celecoxib in heavily pre-treated recurrent ovarian cancer patients. BMC Cancer 2011, 11, 214. [Google Scholar] [CrossRef]
- Young, S.D.; Whissell, M.; Noble, J.C.S.; Cano, P.O.; Lopez, P.G.; Germond, C.J. Phase II Clinical Trial Results Involving Treatment with Low-Dose Daily Oral Cyclophosphamide, Weekly Vinblastine, and Rofecoxib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2006, 12, 3092–3098. [Google Scholar] [CrossRef]
- Ishikawa, H.; Mutoh, M.; Sato, Y.; Doyama, H.; Tajika, M.; Tanaka, S.; Horimatsu, T.; Takeuchi, Y.; Kashida, H.; Tashiro, J.; et al. Chemoprevention with low-dose aspirin, mesalazine, or both in patients with familial adenomatous polyposis without previous colectomy (J-FAPP Study IV): A multicentre, double-blind, randomised, two-by-two factorial design trial. Lancet Gastroenterol. Hepatol. 2021, 6, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Kornmann, M.; Traub, B. Role of Epithelial to Mesenchymal Transition in Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 14815. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Liao, X.; Li, G.; Xiang, J.; Ye, Q.; Chen, M.; Feng, S. The use of non-steroid anti-inflammatory drugs during radical resection correlated with the outcome in non-small cell lung cancer. World J. Surg. Oncol. 2023, 21, 358. [Google Scholar] [CrossRef]
- Pretre, V.; Papadopoulos, D.; Regard, J.; Pelletier, M.; Woo, J. Interleukin-1 (IL-1) and the inflammasome in cancer. Cytokine 2022, 153, 155850. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Ridker, P.; Lorenzatti, A.; Krum, H.; Varigos, J.; et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Xu, Y.; Lou, Z.; Lee, S.-H. Arctigenin represses TGF-β-induced epithelial mesenchymal transition in human lung cancer cells. Biochem. Biophys. Res. Commun. 2017, 493, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Malyla, V.; De Rubis, G.; Paudel, K.R.; Chellappan, D.K.; Hansbro, N.G.; Hansbro, P.M.; Dua, K. Berberine nanostructures attenuate ß-catenin, a key component of epithelial mesenchymal transition in lung adenocarcinoma. Naunyn. Schmiedebergs. Arch. Pharmacol. 2023, 396, 3595–3603. [Google Scholar] [CrossRef]
- Hussain, Y.; Cui, J.H.; Khan, H.; Aschner, M.; Batiha, G.E.-S.; Jeandet, P. Luteolin and cancer metastasis suppression: Focus on the role of epithelial to mesenchymal transition. Med. Oncol. 2021, 38, 66. [Google Scholar] [CrossRef]
- Choudhury, P.; Barua, A.; Roy, A.; Pattanayak, R.; Bhattacharyya, M.; Saha, P. Eugenol emerges as an elixir by targeting β-catenin, the central cancer stem cell regulator in lung carcinogenesis: An in vivo and in vitro rationale. Food Funct. 2021, 12, 1063–1078. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Tang, L.; Chen, H.; Wu, C.; Zhao, M.; Yang, Y.; Chen, X.; Liu, G. Resveratrol inhibits TGF-β1-induced epithelial-to-mesenchymal transition and suppresses lung cancer invasion and metastasis. Toxicology 2013, 303, 139–146. [Google Scholar] [CrossRef]
- Bahrami, A.; Majeed, M.; Sahebkar, A. Curcumin: A potent agent to reverse epithelial-to-mesenchymal transition. Cell. Oncol. 2019, 42, 405–421. [Google Scholar] [CrossRef]
- Ang, H.L.; Mohan, C.D.; Shanmugam, M.K.; Leong, H.C.; Makvandi, P.; Rangappa, K.S.; Bishayee, A.; Kumar, A.P.; Sethi, G. Mechanism of epithelial-mesenchymal transition in cancer and its regulation by natural compounds. Med. Res. Rev. 2023, 43, 1141–1200. [Google Scholar] [CrossRef]
- Anwar, S.; Malik, J.A.; Ahmed, S.; Kameshwar, V.A.; Alanazi, J.; Alamri, A.; Ahemad, N. Can Natural Products Targeting EMT Serve as the Future Anticancer Therapeutics? Molecules 2022, 27, 7668. [Google Scholar] [CrossRef]
- Nan, Y.; Su, H.; Zhou, B.; Liu, S. The function of natural compounds in important anticancer mechanisms. Front. Oncol. 2023, 12, 1049888. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Li, L.; Song, W.; Li, M.; Hua, X.; Wang, Y.; Yuan, J.; Xue, Z. Natural products of pentacyclic triterpenoids: From discovery to heterologous biosynthesis. Nat. Prod. Rep. 2023, 40, 1303–1353. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Li, H.; Zhang, S.; Lu, H.; Chen, Q. A Review on Preparation of Betulinic Acid and Its Biological Activities. Molecules 2021, 26, 5583. [Google Scholar] [CrossRef]
- Gudoityte, E.; Arandarcikaite, O.; Mazeikiene, I.; Bendokas, V.; Liobikas, J. Ursolic and oleanolic acids: Plant metabolites with neuroprotective potential. Int. J. Mol. Sci. 2021, 22, 4599. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-S.; Xia, H.-M.; Wu, C.-H.; Li, C.-B.; Duan, C.-C.; Che, C.; Zhang, X.-J.; Li, H.-T.; Zhang, Y.; Zhang, X.-F. Novel nortriterpenoids with new skeletons and limonoids from the fruits of Evodia rutaecarpa and their bioactivities. Fitoterapia 2020, 142, 104503. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Sun, J.; You, W.; Wei, Y.; Tian, H.; Jiang, S. Glycyrrhizin suppresses epithelial-mesenchymal transition by inhibiting high-mobility group box1 via the TGF-β1/Smad2/3 pathway in lung epithelial cells. PeerJ 2020, 8, e8514. [Google Scholar] [CrossRef] [PubMed]
- Ayeleso, T.B.; Matumba, M.G.; Mukwevho, E. Oleanolic Acid and Its Derivatives: Biological Activities and Therapeutic Potential in Chronic Diseases. Molecules 2017, 22, 1915. [Google Scholar] [CrossRef]
- Li, J.; Guo, Q.; Lei, X.; Zhang, L.; Su, C.; Liu, Y.; Zhou, W.; Chen, H.; Wang, H.; Wang, F.; et al. Pristimerin induces apoptosis and inhibits proliferation, migration in H1299 Lung Cancer Cells. J. Cancer 2020, 11, 6348–6355. [Google Scholar] [CrossRef]
- Kang, H.; Lee, M.; Jang, S.W. Celastrol inhibits TGF-β1-induced epithelial-mesenchymal transition by inhibiting Snail and regulating E-cadherin expression. Biochem. Biophys. Res. Commun. 2013, 437, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Mannino, G.; Iovino, P.; Lauria, A.; Genova, T.; Asteggiano, A.; Notarbartolo, M.; Porcu, A.; Serio, G.; Chinigò, G.; Occhipinti, A.; et al. Bioactive Triterpenes of Protium heptaphyllum Gum Resin Extract Display Cholesterol-Lowering Potential. Int. J. Mol. Sci. 2021, 22, 2664. [Google Scholar] [CrossRef] [PubMed]
- Oboh, M.; Govender, L.; Siwela, M.; Mkhwanazi, B.N. Anti-Diabetic Potential of Plant-Based Pentacyclic Triterpene Derivatives: Progress Made to Improve Efficacy and Bioavailability. Molecules 2021, 26, 7243. [Google Scholar] [CrossRef] [PubMed]
- Colla, A.R.S.; Rosa, J.M.; Cunha, M.P.; Rodrigues, A.L.S. Anxiolytic-like effects of ursolic acid in mice. Eur. J. Pharmacol. 2015, 758, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.L.; Lima-Júnior, R.C.P.; David, J.P.; David, J.M.; Santos, F.A.; Rao, V.S. Oleanolic acid, a pentacyclic triterpene attenuates the mustard oil-induced colonic nociception in mice. Biol. Pharm. Bull. 2006, 29, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.D.S.; de Jesus, B.d.S.M.; da Silva Luiz, S.R.; Viana, C.B.; Adão Malafaia, C.R.; Figueiredo, F.D.S.; Carvalho, T.d.S.C.; Silva, M.L.; Londero, V.S.; da Costa-Silva, T.A.; et al. Antiinflammatory activity of natural triterpenes—An overview from 2006 to 2021. Phyther. Res. 2022, 36, 1459–1506. [Google Scholar] [CrossRef]
- Özdemir, Z.; Wimmer, Z. Selected plant triterpenoids and their amide derivatives in cancer treatment: A review. Phytochemistry 2022, 203, 113340. [Google Scholar] [CrossRef]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-me, two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef]
- Zhu, B.; Wei, Y. Antitumor activity of celastrol by inhibition of proliferation, invasion, and migration in cholangiocarcinoma via PTEN/PI3K/Akt pathway. Cancer Med. 2020, 9, 783–796. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, W.; Zhao, J.; Zhang, H.; Zhang, Q.; Liang, Y.; Chen, S.; Liu, H.; Zong, S.; Tian, Y.; et al. Oleanolic acid inhibits epithelial–mesenchymal transition of hepatocellular carcinoma by promoting iNOS dimerization. Mol. Cancer Ther. 2019, 18, 62–74. [Google Scholar] [CrossRef]
- Cao, M.; Xiao, D.; Ding, X. The anti-tumor effect of ursolic acid on papillary thyroid carcinoma via suppressing Fibronectin-1. Biosci. Biotechnol. Biochem. 2020, 84, 2415–2424. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, P.; Wang, N.; Wang, S.; Yang, B.; Li, M.; Chen, J.; Situ, H.; Xie, M.; Lin, Y.; et al. Betulinic Acid Suppresses Breast Cancer Metastasis by Targeting GRP78-Mediated Glycolysis and ER Stress Apoptotic Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 8781690. [Google Scholar] [CrossRef]
- Cui, Q.; Ren, J.; Zhou, Q.; Yang, Q.; Li, B. Effect of asiatic acid on epithelial-mesenchymal transition of human alveolar epithelium A549 cells induced by TGF-β1. Oncol. Lett. 2019, 17, 4285–4292. [Google Scholar] [CrossRef]
- Dong, S.-H.H.; Liu, Y.-W.W.; Wei, F.; Tan, H.-Z.Z.; Han, Z.-D.D. Asiatic acid ameliorates pulmonary fibrosis induced by bleomycin (BLM) via suppressing pro-fibrotic and inflammatory signaling pathways. Biomed. Pharmacother. 2017, 89, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Guo, L.; Miao, L.; Bao, W.; Yang, J.; Li, X.; Xi, T.; Zhao, W. Ursolic acid inhibits epithelial-mesenchymal transition by suppressing the expression of astrocyte-elevated gene-1 in human nonsmall cell lung cancer A549 cells. Anticancer Drugs 2013, 24, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.S.; Zhou, H.; Yang, L.; Wang, L.; Jiang, Z.S.; Sun, H.; Wang, S.M. Ursolic acid attenuates TGF-b1-induced epithelial-mesenchymal transition in NSCLC by targeting integrin Avb5/MMPs signaling. Oncol. Res. 2019, 27, 593–600. [Google Scholar] [CrossRef]
- Lin, L.; Hou, G.; Han, D.; Yin, Y.; Kang, J.; Wang, Q. Ursolic acid alleviates airway-vessel remodeling and muscle consumption in cigarette smoke-induced emphysema rats. BMC Pulm. Med. 2019, 19, 103. [Google Scholar] [CrossRef]
- Lin, L.; Hou, G.; Han, D.; Kang, J.; Wang, Q. Ursolic Acid Protected Lung of Rats From Damage Induced by Cigarette Smoke Extract. Front. Pharmacol. 2019, 10, 700. [Google Scholar] [CrossRef]
- Kang, D.Y.; Sp, N.; Lee, J.-M.; Jang, K.-J. Antitumor effects of ursolic acid through mediating the inhibition of STAT3/PD-L1 signaling in non-small cell lung cancer cells. Biomedicines 2021, 9, 297. [Google Scholar] [CrossRef]
- Yang, K.; Chen, Y.; Zhou, J.; Ma, L.; Shan, Y.; Cheng, X.; Wang, Y.; Zhang, Z.; Ji, X.; Chen, L.; et al. Ursolic acid promotes apoptosis and mediates transcriptional suppression of CT45A2 gene expression in non-small-cell lung carcinoma harbouring EGFR T790M mutations. Br. J. Pharmacol. 2019, 176, 4609–4624. [Google Scholar] [CrossRef]
- Son, J.; Lee, S.Y. Ursonic acid exerts inhibitory effects on matrix metalloproteinases via ERK signaling pathway. Chem. Biol. Interact. 2020, 315, 108910. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Zhang, C.-Y.; Ma, Y.-Q.; He, Z.-X.; Zhe, H.; Zhou, S.-F. Therapeutic effects of C-28 methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO-Me; bardoxolone methyl) on radiation-induced lung inflammation and fibrosis in mice. Drug Des. Dev. Ther. 2015, 9, 3163–3178. [Google Scholar] [CrossRef]
- Divya, T.; Velavan, B.; Sudhandiran, G. Regulation of Transforming Growth Factor-β/Smad-mediated Epithelial–Mesenchymal Transition by Celastrol Provides Protection against Bleomycin-induced Pulmonary Fibrosis. Basic Clin. Pharmacol. Toxicol. 2018, 123, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhong, Q.; Zhong, R.; Huang, H.; Xia, Z.; Ke, Z.; Zhang, Z.; Song, J.; Jia, X. Preparation and antitumor evaluation of self-assembling oleanolic acid-loaded Pluronic P105/d-α-tocopheryl polyethylene glycol succinate mixed micelles for non-small-cell lung cancer treatment. Int. J. Nanomed. 2016, 11, 6337–6352. [Google Scholar] [CrossRef]
- Markov, A.V.; Odarenko, K.V.; Sen’kova, A.V.; Salomatina, O.V.; Salakhutdinov, N.F.; Zenkova, M.A. Cyano enone-bearing triterpenoid soloxolone methyl inhibits epithelial-mesenchymal transition of human lung adenocarcinoma cells in vitro and metastasis of murine melanoma in vivo. Molecules 2020, 25, 5925. [Google Scholar] [CrossRef] [PubMed]
- Patlolla, J.M.R.; Qian, L.; Biddick, L.; Zhang, Y.; Desai, D.; Amin, S.; Lightfoot, S.; Rao, C. V β-Escin inhibits NNK-induced lung adenocarcinoma and ALDH1A1 and RhoA/Rock expression in A/J mice and growth of H460 human lung cancer cells. Cancer Prev. Res. 2013, 6, 1140–1149. [Google Scholar] [CrossRef]
- Bang, I.J.; Kim, H.R.; Jeon, Y.; Jeong, M.H.; Park, Y.J.; Kwak, J.H.; Chung, K.H. β-Peltoboykinolic Acid from Astilbe rubra Attenuates TGF-β1-Induced Epithelial-to-Mesenchymal Transitions in Lung Alveolar Epithelial Cells. Molecules 2019, 24, 2573. [Google Scholar] [CrossRef] [PubMed]
- He, D.-H.; Chen, Y.-F.; Zhou, Y.-L.; Zhang, S.-B.; Hong, M.; Yu, X.; Wei, S.-F.; Fan, X.-Z.; Li, S.-Y.; Wang, Q.; et al. Phytochemical library screening reveals betulinic acid as a novel Skp2-SCF E3 ligase inhibitor in non–small cell lung cancer. Cancer Sci. 2021, 112, 3218–3232. [Google Scholar] [CrossRef]
- Hsu, T.-I.I.; Chen, Y.-J.J.; Hung, C.-Y.Y.; Wang, Y.-C.C.; Lin, S.-J.J.; Su, W.-C.C.; Lai, M.-D.D.; Kim, S.-Y.Y.; Wang, Q.; Qian, K.; et al. A novel derivative of betulinic acid, SYK023, suppresses lung cancer growth and malignancy. Oncotarget 2015, 6, 13671–13687. [Google Scholar] [CrossRef]
- Pansters, N.A.; Langen, R.C.; Wouters, E.F.; Schols, A.M. Synergistic stimulation of myogenesis by glucocorticoid and IGF-I signaling. J. Appl. Physiol. 2013, 114, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Hance, M.W.; Dole, K.; Gopal, U.; Bohonowych, J.E.; Jezierska-Drutel, A.; Neumann, C.A.; Liu, H.; Garraway, I.P.; Isaacs, J.S. Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J. Biol. Chem. 2012, 287, 37732–37744. [Google Scholar] [CrossRef] [PubMed]
- Salomatina, O.V.; Markov, A.V.; Logashenko, E.B.; Korchagina, D.V.; Zenkova, M.A.; Salakhutdinov, N.F.; Vlassov, V.V.; Tolstikov, G.A. Synthesis of novel 2-cyano substituted glycyrrhetinic acid derivatives as inhibitors of cancer cells growth and NO production in LPS-activated J-774 cells. Bioorg. Med. Chem. 2014, 22, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, K.; Yanagida-Asanuma, E.; Faul, C.; Tomino, Y.; Kim, K.; Mundel, P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat. Cell Biol. 2006, 8, 485–491. [Google Scholar] [CrossRef]
- Zhang, W.; Ren, Z.; Jia, L.; Li, X.; Jia, X.; Han, Y. Fbxw7 and Skp2 regulate stem cell switch between quiescence and mitotic division in lung adenocarcinoma. BioMed Res. Int. 2019, 2019, 9648269. [Google Scholar] [CrossRef] [PubMed]
- Vassalli, G. Aldehyde dehydrogenases: Not just markers, but functional regulators of stem cells. Stem Cells Int. 2019, 2019, 3904645. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PT | Experimental Model | Conditions | Biological Effects *** | Effects on Cell Signalings **** | Refs. | ||
|---|---|---|---|---|---|---|---|
| Type | Name | Conc. * | Time ** | ||||
| Ursane | Asiatic acid (AA) | A549 treated with TGF-β1 (10 ng/mL) | 10–40 | 24 h | ↓ morphological changes (20 μM AA), ↓ migration DD, ↓ invasion DD | ↓ β-catenin DD, ↓ p-GSK-3β DD, ↑ E-cadherin DD, ↓ N-cadherin DD, ↓ vimentin DD, ↓ Snail DD | [164] |
| Pulmonary fibrosis in C57BL/6 mice was induced by single intratracheal administration of bleomycin (3 mg/kg) | 5–20 mg/kg/day (intragastrically) | 21 d | ↓ collagen deposition DD, ↓ histopathological changes in the lungs DD, ↑ pulmonary function DD, ↓ macrophage, neutrophil and lymphocyte infiltration in BALF DD | ↓ collagen I DD, ↓ collagen III DD, ↓ α-SMA DD, ↓ TIMP-1 DD, ↓ vimentin DD, ↑ E-cadherin DD, ↓ TGF-β1 DD, ↓ p-Smad2/3 DD, ↓ p-ERK1/2 DD, ↓ IL-1β DD, ↓ IL-18 DD, ↓ IL-6 DD, ↓ TNF-α DD, ↓ NLRP3 DD, ↓ ASC DD, ↓ pro-Caspase-1 DD, ↓ active Caspase-1 DD | [165] | ||
| Ursane | Ursolic acid (UA) | A549, H1975 | 5–30 | 24 h | ↓ adhesion to Matrigel DD (A549), ↓ migration DD (A549, H1975), ↓ invasion DD (A549, H1975) | ↑ E-cadherin DD (A549), ↓ N-cadherin DD (A549), ↓ vimentin (A549, 20 μM UA); 84 genes regulated by UA (A549, 30 μM UA) were associated with the signaling pathways of TGF-β, ECM-receptors, adherens junctions, Wnt, VEGF, tight junctions, cell adhesion molecules; ↓ AEG-1 DD,K (A549) | [166] |
| A549 treated with TNF-α (5 ng/mL) | 5–20 | 12/24 h with UA, then 12/24 h with TNF-α | ↓ NF-κB p65 subunitK (20 μM UA, 12 h), ↓ AEG-1 DD (24 h) | ||||
| H1975 treated with TGF-β1 (5 ng/mL) | 0.02 | 24 h | ↓ morphological changes, ↓ migration, ↓ invasion | ↑ E-cadherin, ↓ N-cadherin, ↓ MMP-2 catalytic activity, ↓ MMP-9 catalytic activity, ↓ MMP-2, ↓ MMP-9, ↓ integrin αVβ5K | [167] | ||
| HBE treated with 1% cigarette smoke extract (CSE) | 10 | 2 h prior to CSE | ↓ TGF-β1, ↓ p-Smad2/3, ↓ S100A4, ↑ IGF-1 | [168] | |||
| Emphysema in Wistar rats was induced by exposure to cigarette smoke for 30 min, two times a day, 6 days a week, for 3 months | 10–40 mg/kg/day (intragastrically) | 3 mos | ↓ airway-vessel remodeling, ↓ collagen deposition, ↓ mucus secretion | ↓ α-SMA DD, ↓ S100A4, ↓ TGF-β1, ↓ p-Smad2/3, ↑ IGF-1 DD | |||
| Emphysema in SD rats induced by exposure to cigarette smoke for 30 min, two times a day, 6 days a week, for 3 months | 10–40 mg/kg/day (intragastrically) | 3 mos | ↓ p-IRE1, ↓ XBP1 | [169] | |||
| Emphysema in SD rats induced by intraperitoneal injection of CSE on days 1, 8, 15, 21 | 20 mg/kg/day (intragastrically) | 2–4 wk | ↓ airway remodeling | ↓ p-Smad2/3, ↓ p-PERK, ↓ PERK, ↓ p-eIF-2α, ↓ e-IF-2α, ↓ ATF4, ↓ CHOP, ↓ p-IRE1, ↓ ATF6, ↓ active Caspase 12 | |||
| A549, H460 | 10, 20 | 1–14 d | ↓ migration DD, ↓ invasion DD, ↓ tumorsphere formation (20 μM UA; 7 d, 14 d) | ↓ VEGF DD (24 h), ↓ NANOG (tumorpheres; 20 μM UA; 24 h), ↓ OCT4 (tumorpheres; 20 μM UA; 24 h), ↓ SOX2 (tumorpheres; 20 μM UA; 24 h), ↓ pEGFR (24 h), ↓ pJAK2 DD (24 h), ↓ pSTAT3 DD (24 h), ↓ PD-L1 DD (24 h), ↓ MMP2 DD (24 h), ↓ MMP3 DD (24 h), ↓ MMP9 DD (24 h), ↓ VEGF DD (24 h), ↓ the binding of STAT3 to MMP2 and PD-L1 promoters (20 μM UA, 24 h) | [170] | ||
| A549 and H460 treated with EGF (25 ng/mL) | 20 | 1 h with EGF, then 24 h with UA | ↓ pEGFR | ||||
| H1975 harbouring L858R/T790M mutation | 25 | 12–72 h | ↓ migration | ↓ CT45A2K (12 h), ↓ the binding of TCF4 to CT45A2 promotorK (12 h), ↓ TCF4 (48 h), ↓ p-β-catenin (48 h), ↑ p-GSK-3β (48 h), ↓ nuclear translocation of β-catenin | [171] | ||
| subcutaneous injection of H1975 in athymic nude mice | 25 mg/kg/day | 18 d | ↓ CT45A2, ↓ p-β-catenin, ↑ p-GSK-3β, ↓ TCF4 | ||||
| Ursane | Ursonic acid | A549, H1299 | 2.5, 5 | 24 h, 48 h | ↓ invasion (24 h) | ↓ MMP-2 catalytic activity DD (A549, H1299; 48 h), ↓ MMP-9 catalytic activity (H1299, 48 h), ↓ MMP-2 DD (A549, H1299; 48 h), ↓ MMP-9 DD (H1299, 48 h), ↓ RECK DD (A549, 48 h), ↑ RECK (H1299, 48 h), ↓ p-ERK DD (A549, H1299; 24 h), ↓ p-CREB DD (A549, H1299; 24 h) | [172] |
| Oleanane | C DDO-Me | Radiation-induced lung inflammation in C57BL/6 mice was induced by thoracic irradiation with a single dose of 12.5 Gy | 600 ng intragastrically on days -1, 1, 3 and 5 | 3 wk | ↓ inflammatory cells infiltration in BALF, ↓ total protein in BALF, ↓ histopathological changes in the lungs | ↓ IL-6, ↓ TGF-β, ↑ IL-10, ↓ fibronectin, ↓ α-SMA, ↓ collagen I | [173] |
| Radiation-induced pulmonary fibrosis in C57BL/6 mice was induced by thoracic irradiation with a single dose of 22.5 Gy | 600 ng intragastrically on days -1, 1, 3, 5, 7 and 9 | 12 wk | ↓ collagen deposition | ↓ fibronectin, ↓ α-SMA, ↓ collagen I | |||
| Friedelane | Celastrol | A549 treated with TGF-β1 (5 ng/mL) | 1 | 30 min with celastrol, then 24 h–72 h with TGF-β1 | ↓ morphological changes (72 h), ↓ invasion (36 h) | ↑ E-cadherin (72 h), ↓ Snail (24 h) | [152] |
| A549 treated with TGF-β1 (5 ng/mL) | 5 | 24 h | ↑ E-cadherin, ↑ ZO-1, ↓ N-cadherin, ↓ vimentin, ↓ Snail, ↓ Slug | [174] | |||
| Pulmonary fibrosis in Wistar albino rats was induced by single intratracheal administration of bleomycin (3 U/kg) | 5 mg/kg, twice a week | 28 d | ↓ TGF-β1, ↓ p-Smad2/3, ↑ E-cadherin, ↑ claudin, ↓ N-cadherin, ↓ Snail, ↓ Slug, ↓ β-catenin, ↓ Hsp90 | ||||
| Oleanane | Evoditrilone A | A549 | 1–2 | 48 h | ↓ colony formation ability DD, ↓ migration DD | ↑ E-cadherin DD, ↓ MMP-2 DD, ↓ N-cadherin DD | [148] |
| Oleanane | Glycyrrhizin | A549 and BEAS-2B treated with TGF-β1 (5 ng/mL) | 50–200 (A549); 25–100 (BEAS-2B) | 2 h with glycyrrhizin, then 24 h with TGF-β1 | ↓ migration DD | ↓ HMGB1 secretion DD, ↓ HMGB1 DD, ↓ p-Smad2/3 DD, ↑ E-cadherin DD, ↓ vimentin DD | [149] |
| A549 and BEAS-2B with lentivirus-mediated HMGB1 overexpression | 100 (A549); 50 (BEAS-2B) | 24 h | ↓ HMGB1 secretion, ↓ HMGB1, ↓ TGF-β1, ↓ p-Smad2/3, ↑ E-cadherin, ↓ vimentin | ||||
| Oleanane | Oleanolic acid (OA), OA-loaded P105/TPGS mixed micelles | A549, PC-9 | 15, 30 | 24 h | ↓ migration (OA-micelles > free OA), ↓ invasion (OA-micelles > free OA) | ↑ E-cadherin (OA-micelles > free OA), ↓ N-cadherin (OA-micelles > free OA), ↓ p-ERK (OA-micelles > free OA) | [175] |
| Friedelane | Pristimerin (Pr) | H1299 | 0.9–3.6 | 24–72 h | ↓ colony formation ability TD (1.8 μM Pr), ↓ migration TD,DD, ↓ invasion DD (48 h) | ↓ vimentin (3.6 μM Pr, 48 h), ↓ F-actin (0.9–3.6 μM Pr, 48 h), ↓ integrin β1 (3.6 μM Pr, 48 h), ↓ MMP-2 (0.9–3.6 μM Pr, 48 h), ↓ Snail (0.9–3.6 μM Pr, 48 h) | [151] |
| Oleanane | Soloxolone methyl | A549 treated with TGF-β1 (50 ng/mL) | 0.5 | 24, 48 h | ↓ morphological changes, ↓ migration (24 h, 48 h), ↓ invasion (48 h) | ↑ E-cadherin (48 h), ↑ ZO-1 (48 h), ↓ vimentin (48 h), ↓ fibronectin (48 h) | [176] |
| Oleanane | β-escin (β-Es) | H460 | 5–40 | 24 h | ↓ ALDH+ cell population (5–40 μM β-Es), ↑ p21 (20 μM β-Es; both in ALDH+ and ALDH- cells) | [177] | |
| lung tumors in A/J mice were induced by single intraperitoneal injection of tobacco carcinogen NNK (10 μmol/mouse) | 3 weeks after NNK treatment mice were fed with the diet containing 500 ppm β-Es | 17, 34 wk | ↓ lung tumor formation, ↓ progression of adenomas to adenocarcinomas | ↑ p21 (34 wk), ↓ ALDH1A1 (34 wk), ↓ p-Akt (34 wk) | |||
| Oleanane | β-peltoboykinolic acid (β-P) | A549 treated with TGF-β1 (2 ng/mL) | 1–10 μg/mL | 24–48 h | ↓ morphological changes (5 μg/mL β-P, 10 μg/mL β-P; 48 h), ↓ migration DD (24 h, 36 h) | ↑ E-cadherin DD (48 h), ↓ N-cadherin (10 μg/mL β-P, 48 h), ↓ vimentin DD (48 h), ↓ fibronectin DD (48 h), ↓ collagen I DD (48 h), ↓ p-Smad2 (10 μg/mL β-P, 48 h), ↓ Snail DD (48 h) | [178] |
| Lupane | Betulinic acid | 293T, A549, H1299 | 10–30 | 4 h–7 d | ↓ migration (A549, H1299; 20 μM BA > 10 μM BA; 24 h), ↓ invasion (A549, H1299; 20 μM BA > 10 μM BA; 24 h), ↓ the sphere-forming ability (A549, H1299; 20 μM BA > 10 μM BA; 7 d) | the direct binding to Skp2 by forming H-bonds with Lys145, ↓ Skp2-Skp1 interactions (exogenous Flag-Skp1 was transfected in 293T, endogenous Skp2-Skp1 interactions in H1299; 20, 30 μM BA; 4 h), ↓ Skp2-mediated ubiquitination of p27 (exogenous p27 in 293T, endogenous p27 in A549; 10, 20 μM BA; 24 h), ↓ Skp2-mediated ubiquitination of E-cadherin (exogenous E-cadherin in 293T, endogenous E-cadherin in A549; 10, 20 μM BA; 24 h), ↓ Skp2 DD,TD (A549, H1299), ↑ p27 DD,TD (A549, H1299), ↑ E-cadherin DD,TD (A549, H1299) | [179] |
| intravenous injection of A549 into BALB/C nude mice (metastasis model) | 50 or 75 mg/kg BA was administered on the day 7 after LLC injection | 2 mos | ↓ metastasis DD | ||||
| A549 and H1299 treated with TGF-β1 (10 ng/mL) | 10–20 | 24 h | ↑ E-cadherin DD, ↓ vimentin (A549: 20 μM; H1299 DD), ↓ N-cadherin DD, ↓ Skp2 DD | ||||
| LLC | 10–20 | 24 h | ↓ migration DD, ↓ invasion DD | ↓ Skp2 DD, ↑ E-cadherin DD | |||
| subcutaneous injection of LLC into C57BL/6 mice (spontaneous metastasis model) | 50 or 75 mg/kg each day after LLC injection | 21 d | ↓ primary tumor growth DD, ↓ lung metastasis DD | ↓ Skp2 DD, ↑ E-cadherin DD, ↑ p27 DD | |||
| intravenous injection of LLC into C57BL/6 mice (metastasis model) | 50 or 75 mg/kg BA was administered on the day 7 after LLC injection | 60 d | ↓ lung metastasis DD | ↓ Skp2, ↑ E-cadherin | |||
| Lupane | Betulinic acid, SYK023 | H1299 | 0.1–30 | 36 h | ↓ migration (BA: 10 μM > 5 μM; SYK023 DD), ↓ invasion (BA: 10 μM > 5 μM; SYK023 DD) | ↓ F-actin polymerization (BA: 10 μM > 5 μM; SYK023 DD), ↓ p-FAK (SYK023: 1 μM, 5 μM), ↓ p-Src (BA: 5 μM > 1 μM; SYK023: 0.5–5 μM), ↓ p-Akt (BA: 5 μM; SYK023: 0.5–5 μM), ↓ p-mTOR (SYK023: 1 μM, 5 μM), ↓ N-cadherin (BA: 30 μM; SYK023 DD), ↓ β-catenin (BA: 30 μM; SYK023 DD), ↓ vimentin (SYK023: 20 μM, 30 μM), ↓ c-Myc (BA: 20 μM, 30 μM; SYK023: 20 μM, 30 μM)), ↓ SYPD K (BA, SYK023: 20 μM) | [180] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odarenko, K.V.; Zenkova, M.A.; Markov, A.V. The Nexus of Inflammation-Induced Epithelial-Mesenchymal Transition and Lung Cancer Progression: A Roadmap to Pentacyclic Triterpenoid-Based Therapies. Int. J. Mol. Sci. 2023, 24, 17325. https://doi.org/10.3390/ijms242417325
Odarenko KV, Zenkova MA, Markov AV. The Nexus of Inflammation-Induced Epithelial-Mesenchymal Transition and Lung Cancer Progression: A Roadmap to Pentacyclic Triterpenoid-Based Therapies. International Journal of Molecular Sciences. 2023; 24(24):17325. https://doi.org/10.3390/ijms242417325
Chicago/Turabian StyleOdarenko, Kirill V., Marina A. Zenkova, and Andrey V. Markov. 2023. "The Nexus of Inflammation-Induced Epithelial-Mesenchymal Transition and Lung Cancer Progression: A Roadmap to Pentacyclic Triterpenoid-Based Therapies" International Journal of Molecular Sciences 24, no. 24: 17325. https://doi.org/10.3390/ijms242417325