
Eosinophilic Chronic Rhinosinusitis and Pathogenic Role of Protease

 , , and
, , and
Abstract
:1. Introduction
2. Eosinophilic Chronic Rhinosinusitis (eCRS)
2.1. Classification of Chronic Rhinosinusitis
2.2. Clinical Presentation, Diagnosis, and Treatment of eCRS
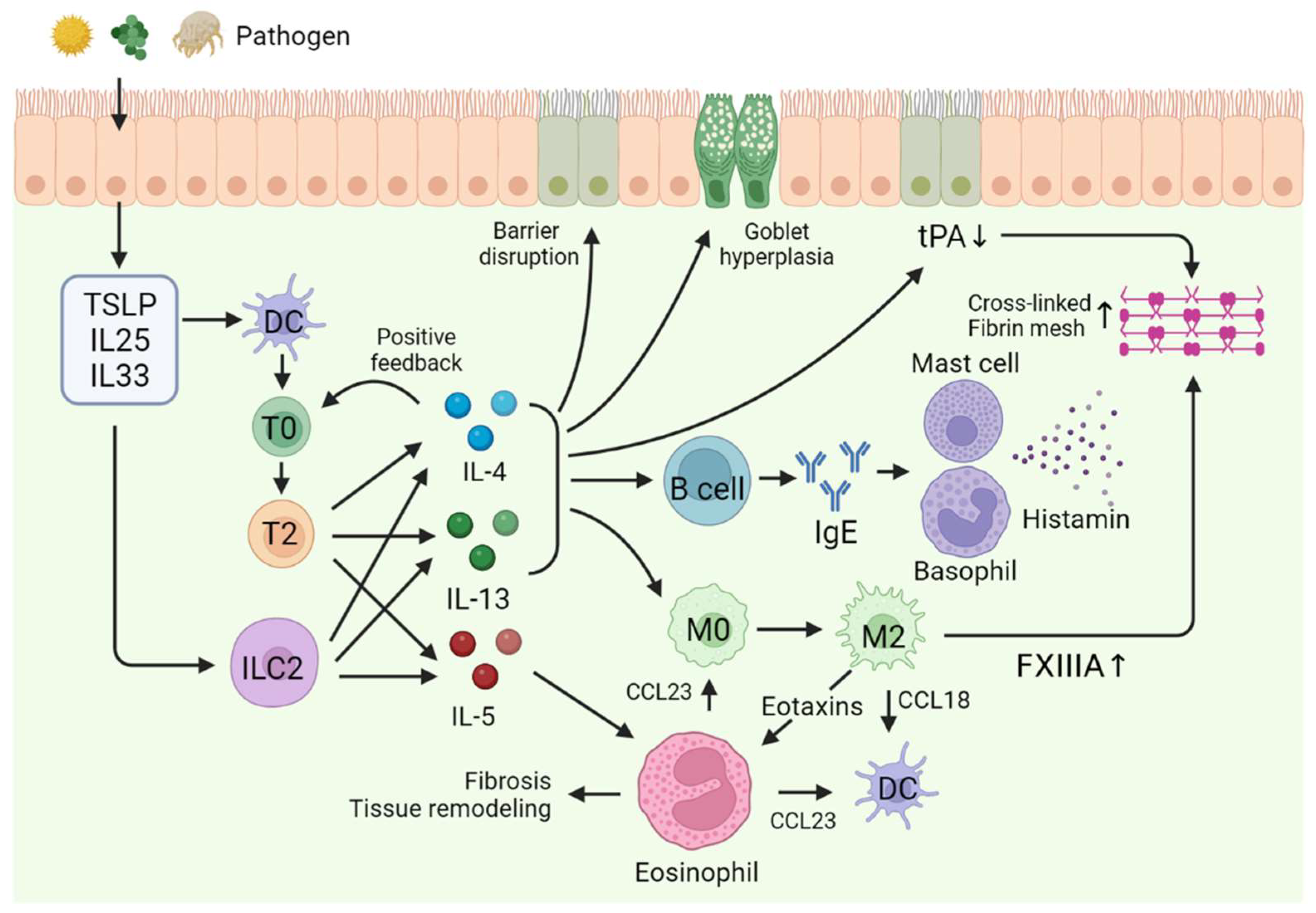
2.3. Pathogenesis of eCRS
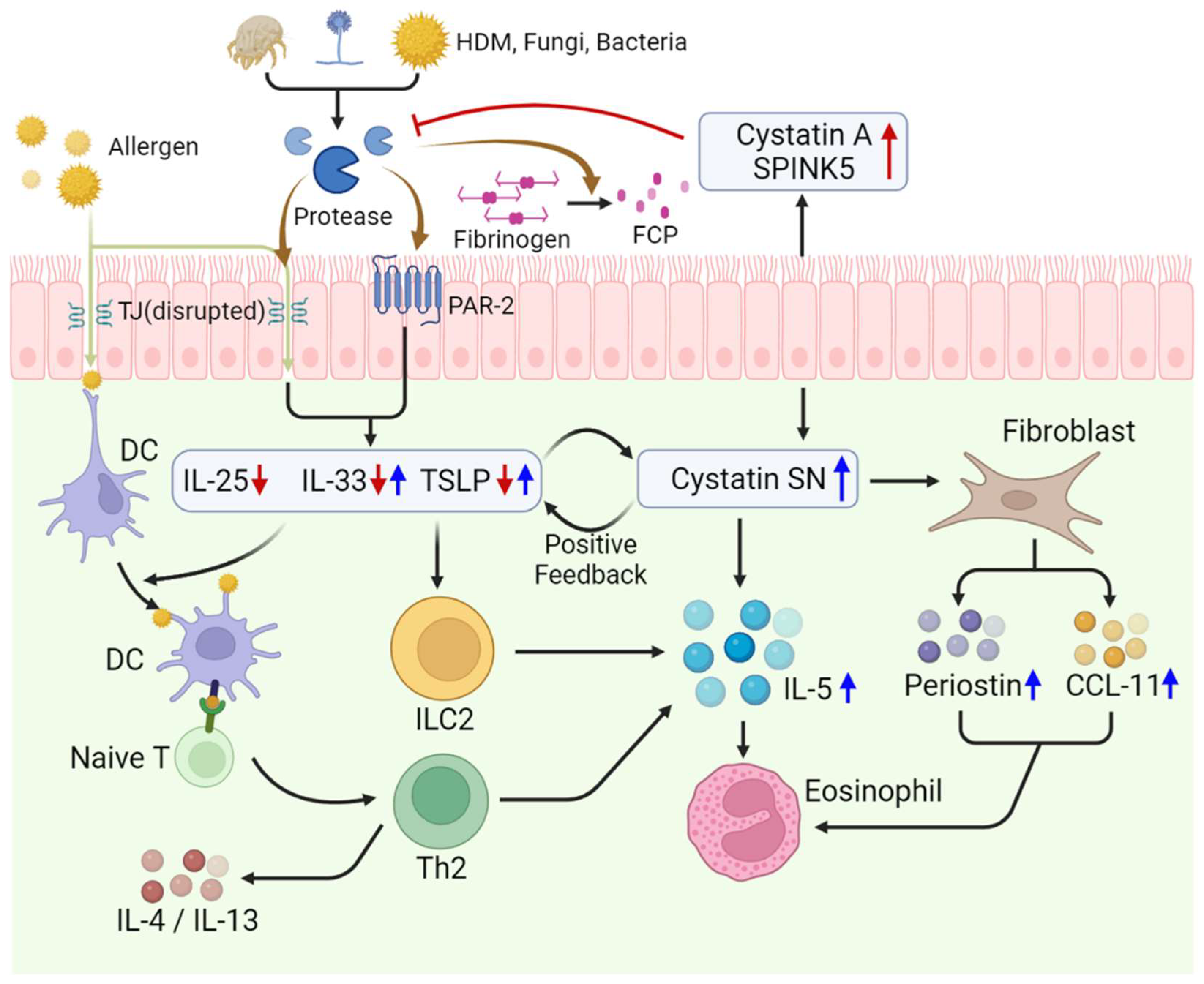
3. Proteases
3.1. Definition and Functions of Protease
3.2. Classification of Proteases
3.3. Role of Proteases in Type 2 Inflammatory Diseases
3.4. Role of Protease Inhibitors in eCRS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fokkens, W.J.; Lund, V.J.; Hopkins, C.; Hellings, P.W.; Kern, R.; Reitsma, S.; Toppila-Salmi, S.; Bernal-Sprekelsen, M.; Mullol, J.; Alobid, I.; et al. European Position Paper on Rhinosinusitis and Nasal Polyps 2020. Rhinology 2020, 58 (Suppl. S29), 1–464. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, T.; Sakashita, M.; Haruna, T.; Asaka, D.; Takeno, S.; Ikeda, H.; Nakayama, T.; Seki, N.; Ito, S.; Murata, J.; et al. Novel Scoring System and Algorithm for Classifying Chronic Rhinosinusitis: The JESREC Study. Allergy 2015, 70, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Stevens, W.W.; Ocampo, C.J.; Berdnikovs, S.; Sakashita, M.; Mahdavinia, M.; Suh, L.; Takabayashi, T.; Norton, J.E.; Hulse, K.E.; Conley, D.B.; et al. Cytokines in Chronic Rhinosinusitis Role in Eosinophilia and Aspirin-Exacerbated Respiratory Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 682–694. [Google Scholar] [CrossRef]
- Krysko, O.; Teufelberger, A.; Van Nevel, S.; Krysko, D.V.; Bachert, C. Protease/Antiprotease Network in Allergy: The Role of Staphylococcus aureus Protease-Like Proteins. Allergy Eur. J. Allergy Clin. Immunol. 2019, 74, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Fokkens, W.J.; Lund, V.J.; Mullol, J.; Bachert, C.; Alobid, I.; Baroody, F.; Cohen, N.; Cervin, A.; Douglas, R.; Gevaert, P.; et al. EPOS 2012: European Position Paper on Rhinosinusitis and Nasal Polyps 2012. A Summary for Otorhinolaryngologists. Rhinology 2012, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Grayson, J.W.; Hopkins, C.; Mori, E.; Senior, B.; Harvey, R.J. Contemporary Classification of Chronic Rhinosinusitis Beyond Polyps vs No Polyps: A Review. JAMA Otolaryngol.–Head Neck Surg. 2020, 146, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Fujieda, S.; Imoto, Y.; Kato, Y.; Ninomiya, T.; Tokunaga, T.; Tsutsumiuchi, T.; Yoshida, K.; Kidoguchi, M.; Takabayashi, T. Eosinophilic Chronic Rhinosinusitis. Allergol. Int. 2019, 68, 403–412. [Google Scholar] [CrossRef]
- Yoshimura, K.; Kawata, R.; Haruna, S.; Moriyama, H.; Hirakawa, K.; Fujieda, S.; Masuyama, K.; Takenaka, H. Clinical Epidemiological Study of 553 Patients with Chronic Rhinosinusitis in Japan. Allergol. Int. 2011, 60, 491–496. [Google Scholar] [CrossRef]
- Sakuma, Y.; Ishitoya, J.; Komatsu, M.; Shiono, O.; Hirama, M.; Yamashita, Y.; Kaneko, T.; Morita, S.; Tsukuda, M. New Clinical Diagnostic Criteria for Eosinophilic Chronic Rhinosinusitis. Auris Nasus Larynx 2011, 38, 583–588. [Google Scholar] [CrossRef]
- Takeno, S.; Hirakawa, K.; Ishino, T. Pathological Mechanisms and Clinical Features of Eosinophilic Chronic Rhinosinusitis in the Japanese Population. Allergol. Int. 2010, 59, 247–256. [Google Scholar] [CrossRef]
- Ishitoya, J.; Sakuma, Y.; Tsukuda, M. Eosinophilic Chronic Rhinosinusitis in Japan. Allergol. Int. 2010, 59, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Mori, E.; Matsuwaki, Y.; Mitsuyama, C.; Okushi, T.; Nakajima, T.; Moriyama, H. Risk Factors for Olfactory Dysfunction in Chronic Rhinosinusitis. Auris Nasus Larynx 2013, 40, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Kountakis, S.E.; Arango, P.; Bradley, D.; Wade, Z.K.; Borish, L. Molecular and Cellular Staging for the Severity of Chronic Rhinosinusitis. Laryngoscope 2004, 114, 1895–1905. [Google Scholar] [CrossRef]
- Ikeda, K.; Shiozawa, A.; Ono, N.; Kusunoki, T.; Hirotsu, M.; Homma, H.; Saitoh, T.; Murata, J. Subclassification of Chronic Rhinosinusitis with Nasal Polyp Based on Eosinophil and Neutrophil. Laryngoscope 2013, 123, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Zheng, Y.; Liu, P.F.; Guo, L.J. Eosinophilic Chronic Rhinosinusitis in East Asians. World J. Clin. Cases 2014, 2, 873–882. [Google Scholar] [CrossRef]
- Bachert, C.; Patou, J.; Van Cauwenberge, P. The Role of Sinus Disease in Asthma. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 29–36. [Google Scholar] [CrossRef]
- Jeong, W.J.; Lee, C.H.; Cho, S.H.; Rhee, C.S. Eosinophilic Allergic Polyp: A Clinically Oriented Concept of Nasal Polyp. Otolaryngol.–Head Neck Surg. 2011, 144, 241–246. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, N.; Bo, M.; Holtappels, G.; Zheng, M.; Lou, H.; Wang, H.; Zhang, L.; Bachert, C. Diversity of TH Cytokine Profiles in Patients with Chronic Rhinosinusitis: A Multicenter Study in Europe, Asia, and Oceania. J. Allergy Clin. Immunol. 2016, 138, 1344–1353. [Google Scholar] [CrossRef]
- Lou, H.; Zhang, N.; Bachert, C.; Zhang, L. Highlights of Eosinophilic Chronic Rhinosinusitis with Nasal Polyps in Definition, Prognosis, and Advancement. In International Forum of Allergy & Rhinology; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2018; Volume 8, pp. 1218–1225. [Google Scholar] [CrossRef]
- Haruna, S.; Nakanishi, M.; Otori, N.; Moriyama, H. Histopathological Features of Nasal Polyps with Asthma Association: An Immunohistochemical Study. Am. J. Rhinol. 2004, 18, 165–172. [Google Scholar] [CrossRef]
- Pirola, F.; Pace, G.M.; Giombi, F.; Heffler, E.; Paoletti, G.; Nappi, E.; Sanità, W.; Giulietti, G.; Giunta, G.; Ferreli, F.; et al. Outcomes of Non-mucosa Sparing Endoscopic Sinus Surgery (Partial Reboot) in Refractory Chronic Rhinosinusitis with Nasal Polyposis: An Academic Hospital Experience. Laryngoscope 2023, 133, 1584–1589. [Google Scholar] [CrossRef]
- Alsharif, S.; Jonstam, K.; van Zele, T.; Gevaert, P.; Holtappels, G.; Bachert, C. Endoscopic Sinus Surgery for Type-2 CRS wNP: An Endotype-Based Retrospective Study. Laryngoscope 2019, 129, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.W.; Ghushchyan, V.H.; Globe, G.; Schatz, M. Oral corticosteroid exposure and adverse effects in asthmatic patients. J. Allergy Clin. Immunol. 2018, 141, 110–116.e7. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Song, Y.; Alonso-Coello, P.; Vogel, Y.; Rocha, C.; Solà, I.; Santero, M.; Akdis, C.A.; Akdis, M.; Canonica, G.W.; et al. Efficacy and Safety of Treatment with BioLogicals for Severe Chronic Rhinosinusitis with Nasal Polyps: A Systematic Review for the EAACI Guidelines. Allergy 2021, 76, 2337–2353. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Han, J.K.; Desrosiers, M.; Hellings, P.W.; Amin, N.; Lee, S.E.; Mullol, J.; Greos, L.S.; Bosso, J.V.; Laidlaw, T.M.; et al. Efficacy and Safety of Dupilumab in Patients with Severe Chronic Rhinosinusitis with Nasal Polyps (Liberty NP SINUS-24 and Liberty NP SINUS-52): Results from Two Multicentre, Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Phase 3 Trials. Lancet 2019, 394, 1638–1650. [Google Scholar] [CrossRef] [PubMed]
- Kariyawasam, H.H.; Chandrasekharan, D.P.; Jacques, T.; Stokes, P.; Dziadzio, M.; Gane, S.B.; Langan, D.; Rimmer, J. Biologic Treatment for Severe Chronic Rhinosinusitis with Nasal Polyps: A Systematic Review and Meta-analysis. Rhinology 2023, 61, 98–107. [Google Scholar] [CrossRef] [PubMed]
- van der Lans, R.J.L.; Hopkins, C.; Senior, B.A.; Lund, V.J.; Reitsma, S. BioLogicals and Endoscopic Sinus Surgery for Severe Uncontrolled Chronic Rhinosinusitis with Nasal Polyps: An Economic Perspective. J. Allergy Clin. Immunol. Pract. 2022, 10, 1454–1461. [Google Scholar] [CrossRef]
- Parasher, A.K.; Gliksman, M.; Segarra, D.; Lin, T.; Rudmik, L.; Quast, T. Economic Evaluation of Dupilumab Versus Endoscopic Sinus Surgery for the Treatment of Chronic Rhinosinusitis with Nasal Polyps. Int. Forum Allergy Rhinol. 2022, 12, 813–820. [Google Scholar] [CrossRef]
- Scangas, G.A.; Wu, A.W.; Ting, J.Y.; Metson, R.; Walgama, E.; Shrime, M.G.; Higgins, T.S. Cost Utility Analysis of Dupilumab Versus Endoscopic Sinus Surgery for Chronic Rhinosinusitis with Nasal Polyps. Laryngoscope 2021, 131, E26–E33. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Galli, S.J. SnapShot: Integrated Type 2 Immune Responses. Immunity 2015, 43, 408.e1. [Google Scholar] [CrossRef]
- Schleimer, R.P.; Berdnikovs, S. Etiology of Epithelial Barrier Dysfunction in Patients with Type 2 Inflammatory Diseases. J. Allergy Clin. Immunol. 2017, 139, 1752–1761. [Google Scholar] [CrossRef]
- Bartemes, K.R.; Kita, H. Dynamic Role of Epithelium-Derived Cytokines in Asthma. Clin. Immunol. 2012, 143, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Hull, L.; Imrie, A.; Snidvongs, K.; Chin, D.; Pratt, E.; Kalish, L.; Sacks, R.; Earls, P.; Sewell, W.; et al. Interleukin-25 and Interleukin-33 as Mediators of Eosinophilic Inflammation in Chronic Rhinosinusitis. Am. J. Rhinol. Allergy 2015, 29, 175–181. [Google Scholar] [CrossRef]
- Ouyang, Y.; Fan, E.; Li, Y.; Wang, X.; Zhang, L. Clinical Characteristics and Expression of Thymic Stromal Lymphopoetin in Eosinophilic and Non-eosinophilic Chronic Rhinosinusitis. ORL J. Otorhinolaryngol. Relat. Spec. 2013, 75, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. T Helper 2 (Th2) Cell Differentiation, Type 2 Innate Lymphoid Cell (ILC2) Development and Regulation of Interleukin-4 (IL-4) and IL-13 Production. Cytokine 2015, 75, 14–24. [Google Scholar] [CrossRef]
- Maspero, J.; Adir, Y.; Al-Ahmad, M.; Celis-Preciado, C.A.; Colodenco, F.D.; Giavina-Bianchi, P.; Lababidi, H.; Ledanois, O.; Mahoub, B.; Perng, D.W.; et al. Type 2 Inflammation in Asthma and Other Airway Diseases. ERJ Open Res. 2022, 8. Available online: https://openres.ersjournals.com/content/8/3/00576-2021 (accessed on 1 August 2022). [CrossRef] [PubMed]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2018.00888 (accessed on 7 June 2018). [CrossRef]
- Saatian, B.; Rezaee, F.; Desando, S.; Emo, J.; Chapman, T.; Knowlden, S.; Georas, S.N. Interleukin-4 and Interleukin-13 Cause Barrier Dysfunction in Human Airway Epithelial Cells. Tissue Barriers 2013, 1, e24333. [Google Scholar] [CrossRef]
- Kondo, M.; Tamaoki, J.; Takeyama, K.; Nakata, J.; Nagai, A. Interleukin-13 Induces Goblet Cell Differentiation in Primary Cell Culture from Guinea Pig Tracheal Epithelium. Am. J. Respir. Cell Mol. Biol. 2002, 27, 536–541. [Google Scholar] [CrossRef]
- Kim, D.H.; Chu, H.S.; Lee, J.Y.; Hwang, S.J.; Lee, S.H.; Lee, H.M. Up-Regulation of MUC5AC and MUC5B Mucin Genes in Chronic Rhinosinusitis. Arch. Otolaryngol.–Head Neck Surg. 2004, 130, 747–752. [Google Scholar] [CrossRef]
- Qin, Y.; Jiang, Y.; Sheikh, A.S.; Shen, S.; Liu, J.; Jiang, D. Interleukin-13 Stimulates MUC5AC Expression via a STAT6-TMEM16A-ERK1/2 Pathway in Human Airway Epithelial Cells. Int. Immunopharmacol. 2016, 40, 106–114. [Google Scholar] [CrossRef]
- McCormick, S.M.; Heller, N.M. Commentary: Il-4 and IL-13 Receptors and Signaling. Cytokine 2015, 75, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, T.; Kato, A.; Peters, A.T.; Hulse, K.E.; Suh, L.A.; Carter, R.; Norton, J.; Grammer, L.C.; Tan, B.K.; Chandra, R.K.; et al. Increased Expression of Factor XIII-A in Patients with Chronic Rhinosinusitis with Nasal Polyps. J. Allergy Clin. Immunol. 2013, 132, 584–592.e4. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, T.; Kato, A.; Peters, A.T.; Hulse, K.E.; Suh, L.A.; Carter, R.; Norton, J.; Grammer, L.C.; Cho, S.H.; Tan, B.K.; et al. Excessive Fibrin Deposition in Nasal Polyps Caused by Fibrinolytic Impairment Through Reduction of Tissue Plasminogen Activator Expression. Am. J. Respir. Crit. Care Med. 2013, 187, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, T.; Schleimer, R.P. Formation of Nasal Polyps: The Roles of Innate Type 2 Inflammation and Deposition of Fibrin. J. Allergy Clin. Immunol. 2020, 145, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Suzuki, M.; Kato, Y.; Kidoguchi, M.; Kumai, T.; Fujieda, S.; Sakashita, M. The Current Findings in Eosinophilic Chronic Rhinosinusitis. Auris Nasus Larynx 2023. Available online: https://www.sciencedirect.com/science/article/pii/S0385814623001487 (accessed on 11 August 2023).
- Tsuda, T.; Maeda, Y.; Nishide, M.; Koyama, S.; Hayama, Y.; Nojima, S.; Takamatsu, H.; Okuzaki, D.; Kinehara, Y.; Kato, Y.; et al. Eosinophil-Derived Neurotoxin Enhances Airway Remodeling in Eosinophilic Chronic Rhinosinusitis and Correlates with Disease Severity. Int. Immunol. 2019, 31, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, P.; Han, J.K.; Smith, S.G.; Sousa, A.R.; Howarth, P.H.; Yancey, S.W.; Chan, R.; Bachert, C. The Roles of Eosinophils and Interleukin-5 in the Pathophysiology of Chronic Rhinosinusitis with Nasal Polyps. Int. Forum Allergy Rhinol. 2022, 12, 1413–1423. [Google Scholar] [CrossRef]
- Kato, A. Immunopathology of Chronic Rhinosinusitis. Allergol. Int. 2015, 64, 121–130. [Google Scholar] [CrossRef]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional Enzymes in Life and Disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef]
- Barrett, A.J. Proteases. Curr. Protoc. Protein Sci. 2000, 21. Available online: https://currentprotocols.onlinelibrary.wiley.com/doi/10.1002/0471140864.ps2101s21 (accessed on 1 May 2000). [CrossRef]
- Levene, P.A. The Cleavage Products of Proteoses. J. Biol. Chem. 1905, 1, 45–58. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Otto, H.H.; Schirmeister, T. Cysteine Proteases and Their Inhibitors. Chem. Rev. 1997, 97, 133–172. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine Proteases: Modes of Activation and Future Prospects as Pharmacological Targets. Front. Pharmacol. 2016, 7, 193290. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wong, R.N.S. Evolution in the Structure and Function of Aspartic Proteases. J. Cell. Biochem. 1987, 33, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Werb, Z. The Many Faces of Metalloproteases: Cell Growth, Invasion, Angiogenesis and Metastasis. Trends Cell Biol. 2001, 11, S37–S43. [Google Scholar] [CrossRef]
- Klein, T.; Bischoff, R. Physiology and Pathophysiology of Matrix Metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef]
- Sims, A.H.; Dunn-Coleman, N.S.; Robson, G.D.; Oliver, S.G. Glutamic Protease Distribution Is Limited to Filamentous Fungi. FEMS Microbiol. Lett. 2004, 239, 95–101. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. Asparagine Peptide Lyases: A Seventh Catalytic Type of Proteolytic Enzymes. J. Biol. Chem. 2011, 286, 38321–38328. [Google Scholar] [CrossRef]
- Takai, T.; Ikeda, S. Barrier Dysfunction Caused by Environmental Proteases in the Pathogenesis of Allergic Diseases. Allergol. Int. 2011, 60, 25–35. [Google Scholar] [CrossRef]
- Kale, S.L.; Agrawal, K.; Gaur, S.N.; Arora, N. Cockroach Protease Allergen Induces Allergic Airway Inflammation via Epithelial Cell Activation. Sci. Rep. 2017, 7, 42341. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, A.; Yoshimoto, T. Barrier Dysfunction in the Nasal Allergy. Allergol. Int. 2018, 67, 18–23. [Google Scholar] [CrossRef]
- Cocks, T.M.; Moffatt, J.D. Protease-Activated Receptor-2 (PAR2) in the Airways. Pulm. Pharmacol. Ther. 2001, 14, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.E.; Kita, H. The Role of Protease Activation of Inflammation in Allergic Respiratory Diseases. J. Allergy Clin. Immunol. 2004, 114, 997–1008; quiz 1009. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, D.M.; Schuepbach, R.A. Protease-Activated Receptors (PARs): Mechanisms of Action and Potential Therapeutic Modulators in PAR-Driven Inflammatory Diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Takai, T.; Fujimura, T.; Matsuoka, H.; Ogawa, T.; Murayama, K.; Ishii, A.; Ikeda, S.; Okumura, K.; Ogawa, H. Mite Serine Protease Activates Protease-Activated Receptor-2 and Induces Cytokine Release in Human Keratinocytes. Allergy 2009, 64, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Millien, V.O.; Lu, W.; Shaw, J.; Yuan, X.; Mak, G.; Roberts, L.; Song, L.Z.; Knight, J.M.; Creighton, C.J.; Luong, A.; et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science 2013, 341, 792–796. [Google Scholar] [CrossRef]
- Cho, M.; Lee, J.E.; Lim, H.; Shin, H.W.; Khalmuratova, R.; Choi, G.; Kim, H.S.; Choi, W.S.; Park, Y.J.; Shim, I.; et al. Fibrinogen Cleavage Products and Toll-Like Receptor 4 Promote the Generation of Programmed Cell Death 1 Ligand 2–Positive Dendritic Cells in Allergic Asthma. J. Allergy Clin. Immunol. 2018, 142, 530–541.e6. [Google Scholar] [CrossRef]
- Kouzaki, H.; Matsumoto, K.; Kikuoka, H.; Kato, T.; Tojima, I.; Shimizu, S.; Kita, H.; Shimizu, T. Endogenous Protease Inhibitors in Airway Epithelial Cells Contribute to Eosinophilic Chronic Rhinosinusitis. Am. J. Respir. Crit. Care Med. 2017, 195, 737–747. [Google Scholar] [CrossRef]
- Imoto, Y.; Tokunaga, T.; Matsumoto, Y.; Hamada, Y.; Ono, M.; Yamada, T.; Ito, Y.; Arinami, T.; Okano, M.; Noguchi, E.; et al. Cystatin SN Upregulation in Patients with Seasonal Allergic Rhinitis. PLoS ONE 2013, 8, e67057. [Google Scholar] [CrossRef]
- Fukuoka, A.; Matsushita, K.; Morikawa, T.; Adachi, T.; Yasuda, K.; Kiyonari, H.; Fujieda, S.; Yoshimoto, T. Human Cystatin SN Is an Endogenous Protease Inhibitor That Prevents Allergic Rhinitis. J. Allergy Clin. Immunol. 2019, 143, 1153–1162.e12. [Google Scholar] [CrossRef] [PubMed]
- Nocera, A.L.; Mueller, S.K.; Workman, A.D.; Wu, D.; McDonnell, K.; Sadow, P.M.; Amiji, M.M.; Bleier, B.S. Cystatin SN Is a Potent Upstream Initiator of Epithelial-Derived Type 2 Inflammation in Chronic Rhinosinusitis. J. Allergy Clin. Immunol. 2022, 150, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Takabayashi, T.; Sakashita, M.; Imoto, Y.; Tokunaga, T.; Ninomiya, T.; Morikawa, T.; Yoshida, K.; Noguchi, E.; Fujieda, S. Expression and Functional Analysis of CST1 in Intractable Nasal Polyps. Am. J. Respir. Cell Mol. Biol. 2018, 59, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Lou, H.; Wang, Y.; Li, Y.; Meng, Y.; Qi, S.; Wang, M.; Xiao, L.; Wang, C.; Zhang, L. Epithelium-Derived Cystatin SN Enhances Eosinophil Activation and Infiltration Through IL-5 in Patients with Chronic Rhinosinusitis with Nasal Polyps. J. Allergy Clin. Immunol. 2019, 144, 455–469. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Class | Catalytic Residue | Examples | Functions |
|---|---|---|---|
| Serine Proteases | Serine | Trypsin Chymotrypsin Elastase Protein C | Digestion Blood clotting Immune response |
| Cysteine Proteases | Cysteine | Papain Cathepsins Caspases | Senescence Apoptosis Immune response |
| Aspartic Proteases | Aspartic acid | Pepsin Renin HIV-1 Protease | Digestion HIV maturation |
| Metalloproteases | Zinc | Matrix metalloproteinases (MMPs) Aminopeptidase | Tissue remodeling Wound healing Cell signaling |
| Threonine Proteases | Threonine | Proteasome enzymes | Intracellular protein degradation |
| Glutamic Acid Proteases | Glutamic acid | Scytalidoglutamic peptidase Aspergilloglutamic peptidase | Mainly found in pathogenic fungi |
| Asparagine Peptide Lyases | Asparagine | Viral capsid proteins Autotransporters of pathogenic bacteria | Mainly found in bacteria, virus |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Kwak, S.; Lee, J.; Park, I.-H.; Lee, S.H.; Shin, J.M.; Kim, T.H. Eosinophilic Chronic Rhinosinusitis and Pathogenic Role of Protease. Int. J. Mol. Sci. 2023, 24, 17372. https://doi.org/10.3390/ijms242417372
Kim J, Kwak S, Lee J, Park I-H, Lee SH, Shin JM, Kim TH. Eosinophilic Chronic Rhinosinusitis and Pathogenic Role of Protease. International Journal of Molecular Sciences. 2023; 24(24):17372. https://doi.org/10.3390/ijms242417372
Chicago/Turabian StyleKim, Jaehyeong, Sooun Kwak, Juhyun Lee, Il-Ho Park, Seung Hoon Lee, Jae Min Shin, and Tae Hoon Kim. 2023. "Eosinophilic Chronic Rhinosinusitis and Pathogenic Role of Protease" International Journal of Molecular Sciences 24, no. 24: 17372. https://doi.org/10.3390/ijms242417372
APA StyleKim, J., Kwak, S., Lee, J., Park, I.-H., Lee, S. H., Shin, J. M., & Kim, T. H. (2023). Eosinophilic Chronic Rhinosinusitis and Pathogenic Role of Protease. International Journal of Molecular Sciences, 24(24), 17372. https://doi.org/10.3390/ijms242417372