
Neutrophil Elastase Degrades Histone Deacetylases and Sirtuin 1 in Primary Human Monocyte Derived Macrophages



Abstract
1. Introduction
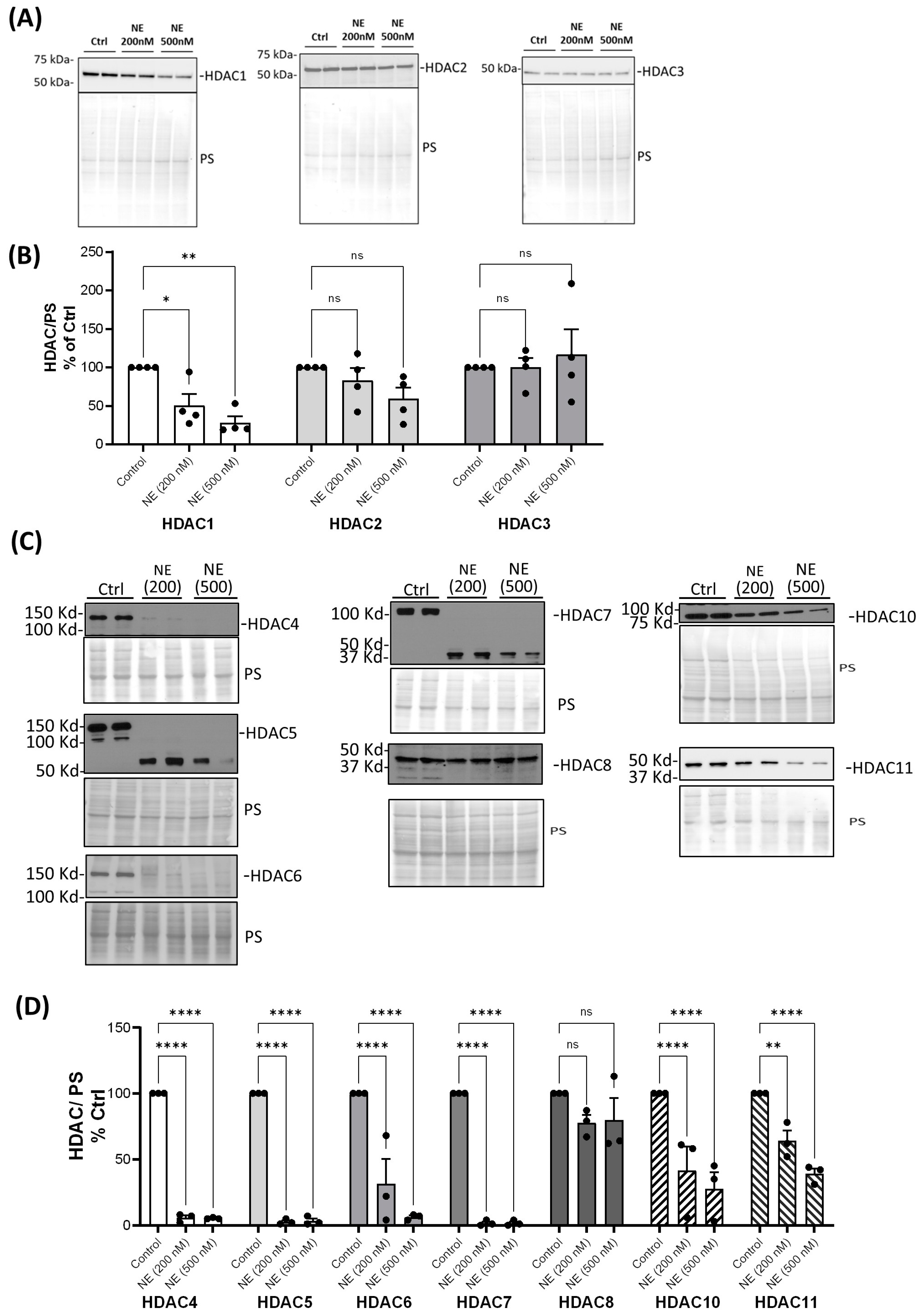
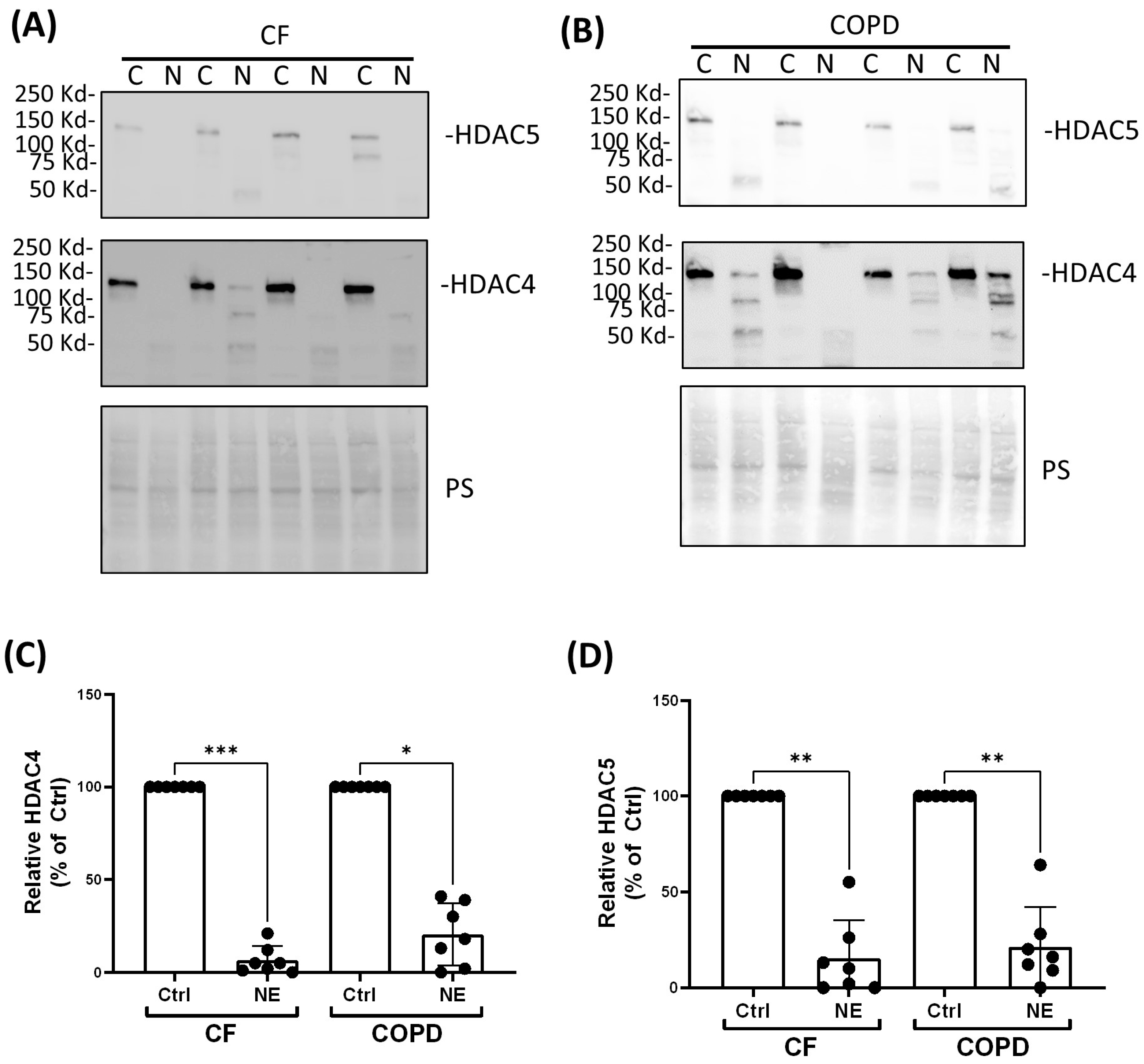
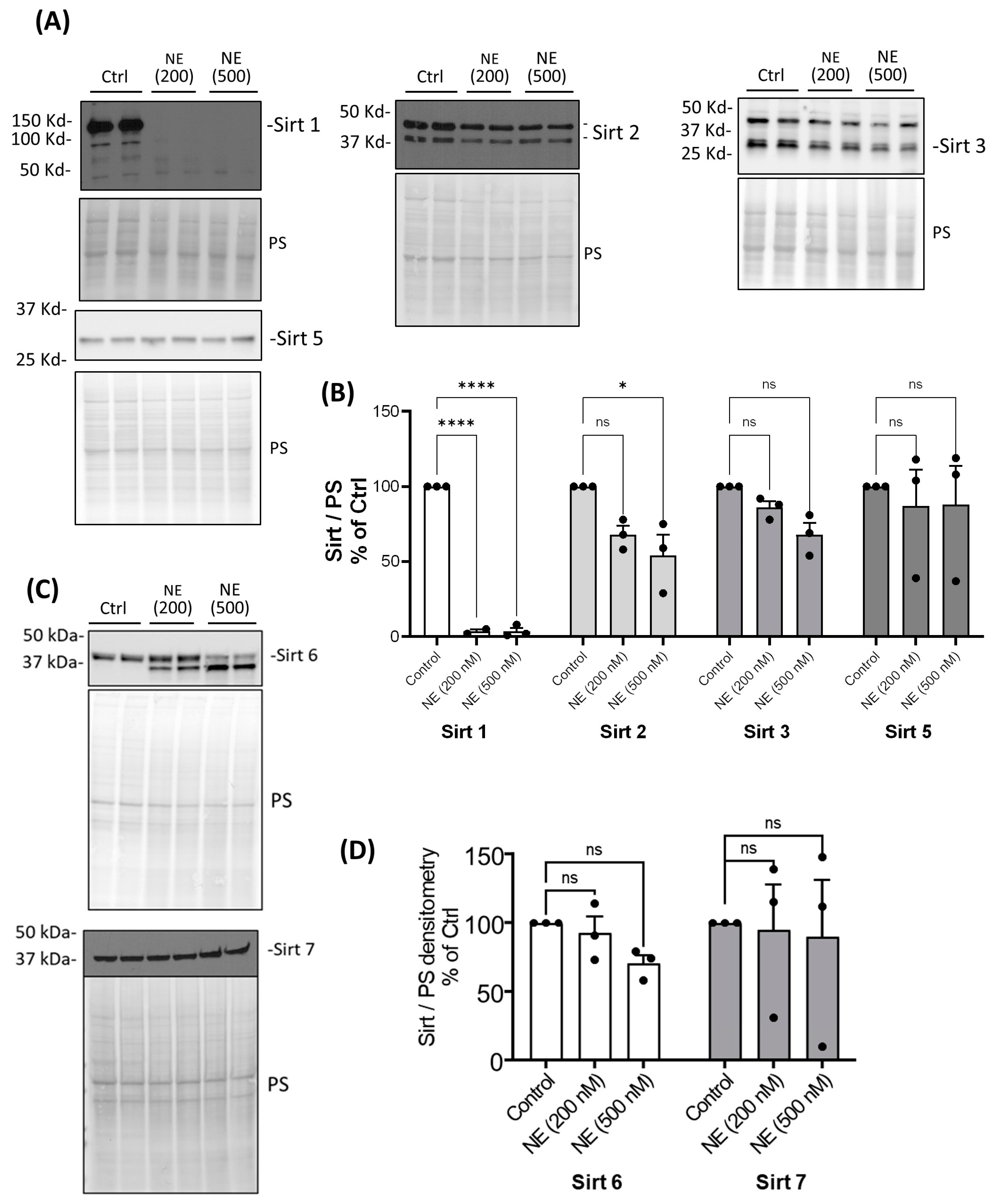
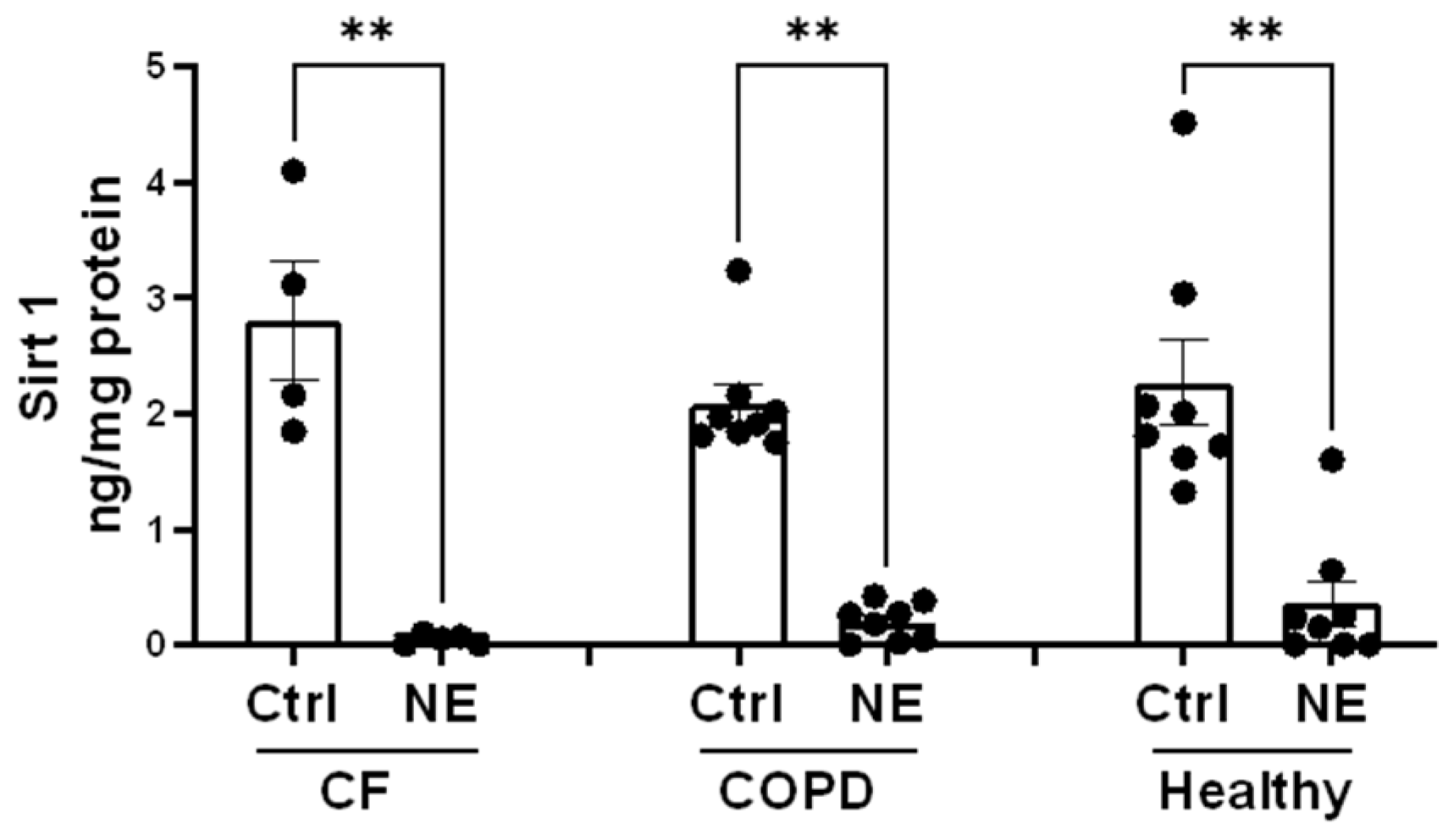
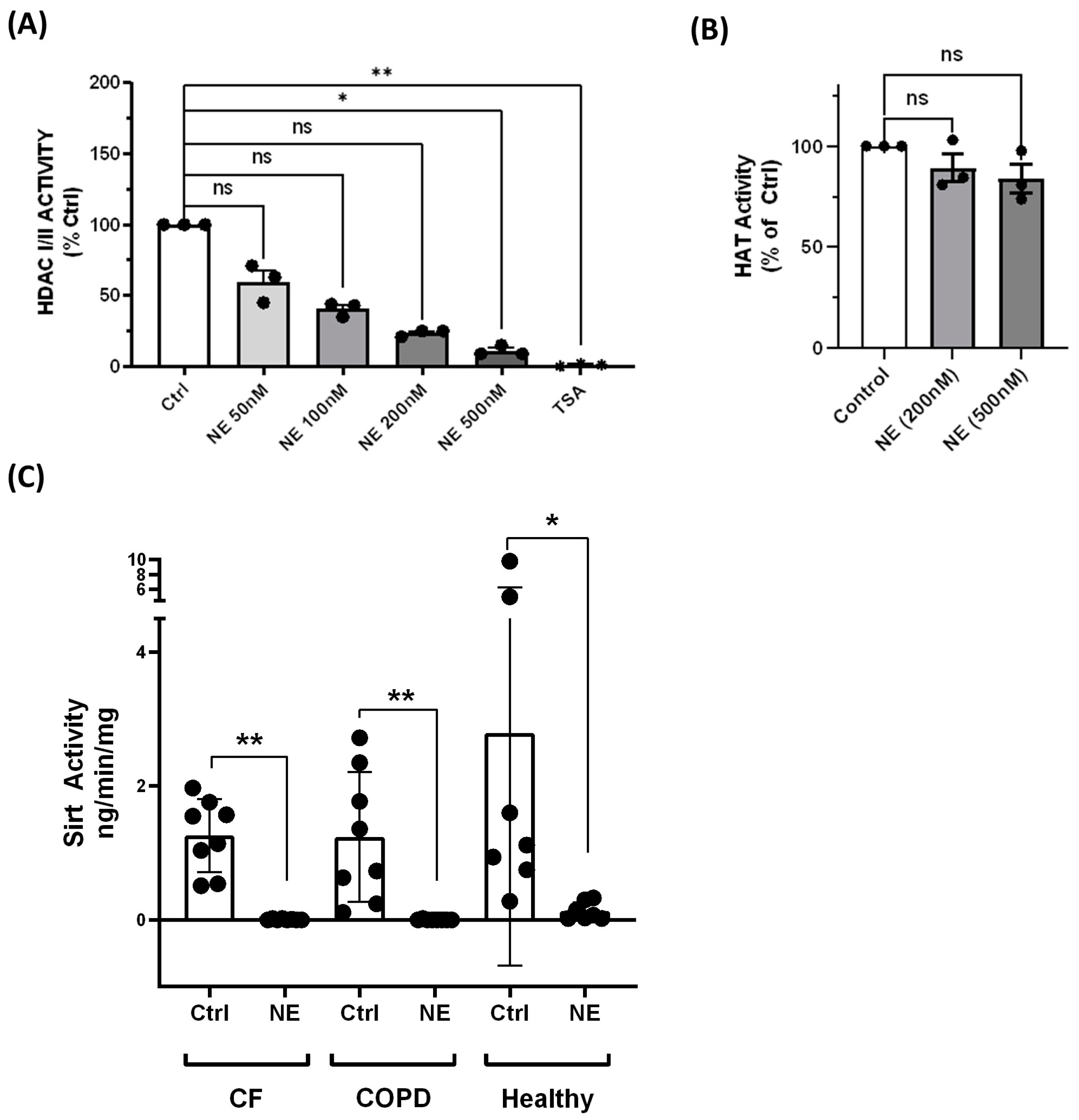
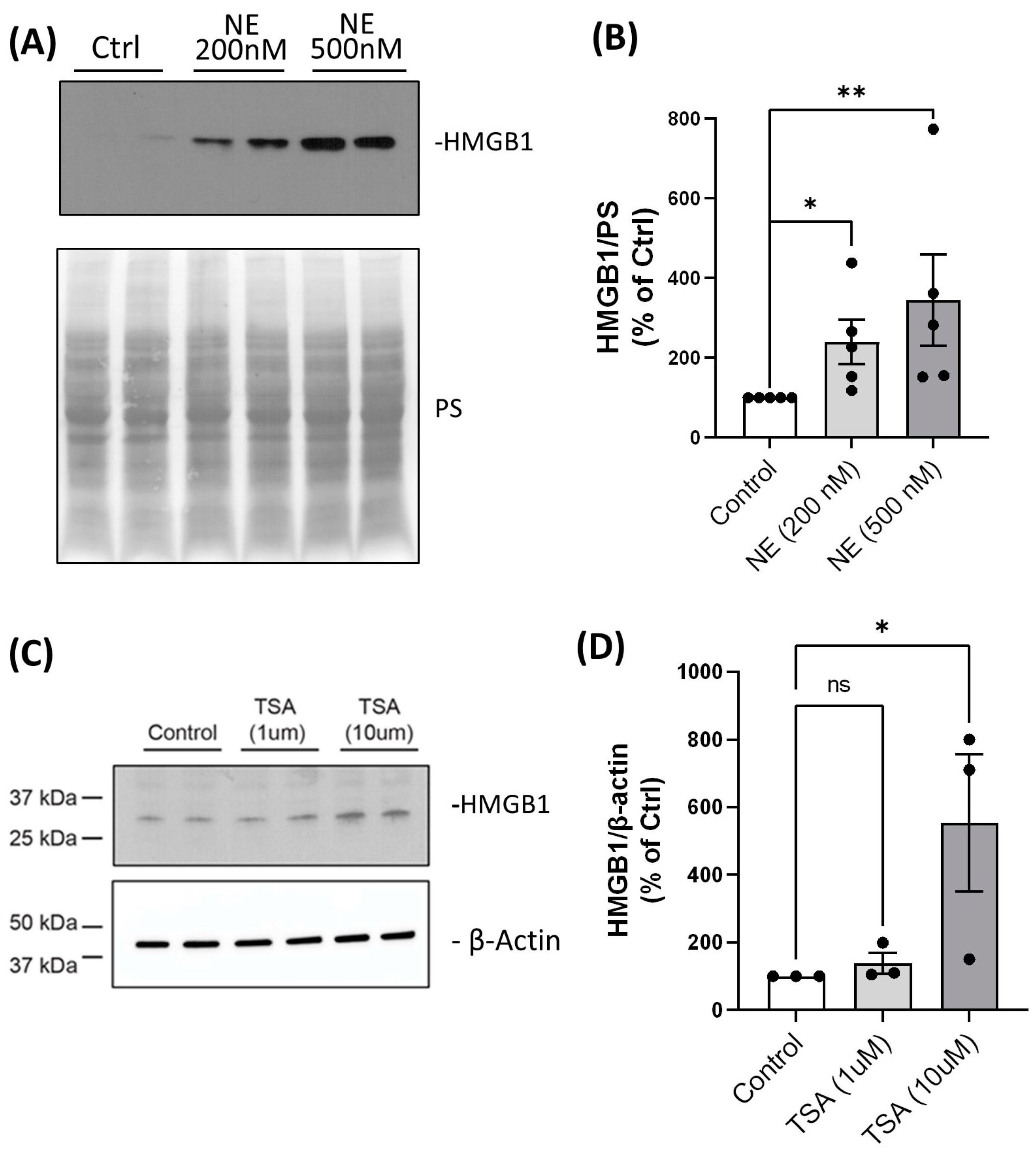
2. Results
3. Discussion
4. Materials and Methods
4.1. Human Subjects
4.2. Buffy Coat Processing Using Double Density Gradients for Human Peripheral Blood Monocyte-Derived Macrophage (BMDM) Cultures
4.3. Monocyte Enrichment via Rosette-Sep Using Buffy Coat and Patient Whole Blood
4.4. Treatments of BMDMs with NE and Trichostatin A (TSA); Total Cell Lysate Collection and Cell Fractionation
4.5. Western Blotting and Antibodies
4.6. HDAC Activity Assays
4.7. HAT Activity Assay
4.8. Sirt 1 ELISA
4.9. Sirt Activity Assay
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Voynow, J.A.; Shinbashi, M. Neutrophil Elastase and Chronic Lung Disease. Biomolecules 2021, 11, 1065. [Google Scholar] [CrossRef] [PubMed]
- Bruscia, E.M.; Bonfield, T.L. Innate and Adaptive Immunity in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Kummarapurugu, A.B.; Zheng, S.; Ma, J.; Ghosh, S.; Hawkridge, A.; Voynow, J.A. Neutrophil Elastase Triggers the Release of Macrophage Extracellular Traps: Relevance to Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 66, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Kummarapurugu, A.B.; Bulut, G.B.; Syed, A.; Kang, L.; Voynow, J.A. Neutrophil elastase activates the release of extracellular traps from COPD blood monocyte-derived macrophages. Clin. Transl. Sci. 2023, 16, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.L.; Xu, X.; Wilson, L.; Weathington, N.M.; Clancy, J.P.; Blalock, J.E.; Gaggar, A. Human Neutrophil Elastase-Mediated Cleavage Sites of MMP-9 and TIMP-1: Implications to Cystic Fibrosis Proteolytic Dysfunction. Mol. Med. 2010, 16, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhan, L.; Li, X.; Yang, Z.; Luo, Y.; Zhao, H. Preclinical and clinical progress for HDAC as a putative target for epigenetic remodeling and functionality of immune cells. Int. J. Biol. Sci. 2021, 17, 3381–3400. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, A.J.; Gennip, A.H.; Caron, H.N.; Kemp, S.; Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased Histone Deacetylase Activity in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, S.; Papaioannou, A.I.; Papaporfyriou, A.; Baker, J.R.; Vuppusetty, C.; Loukides, S.; Barnes, P.J.; Ito, K. Decreased Serum Sirtuin-1 in COPD. Chest 2017, 152, 343–352. [Google Scholar] [CrossRef]
- Wei, L.; Zhang, W.; Li, Y.; Zhai, J. The SIRT1-HMGB1 axis: Therapeutic potential to ameliorate inflammatory responses and tumor occurrence. Front. Cell Dev. Biol. 2022, 10, 986511. [Google Scholar] [CrossRef]
- Zheng, S.; Kummarapurugu, A.B.; Afosah, D.K.; Sankaranarayanan, N.V.; Boothello, R.S.; Desai, U.R.; Kennedy, T.; Voynow, J.A. 2-O, 3-O Desulfated Heparin Blocks High Mobility Group Box 1 Release by Inhibition of p300 Acetyltransferase Activity. Am. J. Respir. Cell Mol. Biol. 2017, 56, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Evankovich, J.; Cho, S.W.; Zhang, R.; Cardinal, J.; Dhupar, R.; Zhang, L.; Klune, J.R.; Zlotnicki, J.; Billiar, T.; Tsung, A. High Mobility Group Box 1 Release from Hepatocytes during Ischemia and Reperfusion Injury Is Mediated by Decreased Histone Deacetylase Activity. J. Biol. Chem. 2010, 285, 39888–39897. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, B.; Wei, X.; Wang, Z.; Fan, B.; Du, P.; Zhang, Y.; Jian, W.; Chen, L.; Wang, L.; et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J. Cell. Mol. Med. 2013, 17, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Rabadi, M.M.; Xavier, S.; Vasko, R.; Kaur, K.; Goligorksy, M.S.; Ratliff, B.B. High-mobility group box 1 is a novel deacetylation target of Sirtuin1. Kidney Int. 2015, 87, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Choi, H.S.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Seo, H.G. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci. Rep. 2015, 5, 15971. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gauthier, A.; Daley, L.; Dial, K.; Wu, J.; Woo, J.; Lin, M.; Ashby, C.; Mantell, L.L. The Role of HMGB1, a Nuclear Damage-Associated Molecular Pattern Molecule, in the Pathogenesis of Lung Diseases. Antioxid. Redox Signal. 2019, 31, 954–993. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Wang, Y.; Zhou, L.; Xiong, J.; Tian, J.; Yang, X.; Gai, X.; Sun, Y. Cigarette smoke-induced HMGB1 translocation and release contribute to migration and NF-kappaB activation through inducing autophagy in lung macrophages. J. Cell. Mol. Med. 2020, 24, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xie, W.; Wang, Y.; Chen, S.; Han, J.; Wang, L.; Gui, P.; Wu, Q. JAK2/STAT1-mediated HMGB1 translocation increases inflammation and cell death in a ventilator-induced lung injury model. Lab. Investig. 2019, 99, 1810–1821. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef]
- Winkler, A.R.; Nocka, K.N.; Williams, C.M. Smoke exposure of human macrophages reduces HDAC3 activity, resulting in enhanced inflammatory cytokine production. Pulm. Pharmacol. Ther. 2012, 25, 286–292. [Google Scholar] [CrossRef]
- Conti, V.; Corbi, G.; Manzo, V.; Malangone, P.; Vitale, C.; Maglio, A.; Cotugno, R.; Capaccio, D.; Marino, L.; Selleri, C.; et al. SIRT1 Activity in Peripheral Blood Mononuclear Cells Correlates with Altered Lung Function in Patients with Chronic Obstructive Pulmonary Disease. Oxidative Med. Cell. Longev. 2018, 2018, 9391261. [Google Scholar] [CrossRef] [PubMed]
- Bartling, T.R.; Drumm, M.L.; Rymut, S.M.; Harker, A.; Corey, D.A.; Burgess, J.D.; Sun, H.; Clancy, J.P.; Kelley, T.J. Loss of CFTR results in reduction of histone deacetylase 2 in airway epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L35–L43. [Google Scholar] [CrossRef] [PubMed]
- Thulborn, S.J.; Mistry, V.; Brightling, C.E.; Moffitt, K.L.; Ribeiro, D.; Bafadhel, M. Neutrophil elastase as a biomarker for bacterial infection in COPD. Respir. Res. 2019, 20, 170. [Google Scholar] [CrossRef] [PubMed]
- Karandashova, S.; Kummarapurugu, A.; Zheng, S.; Kang, L.; Sun, S.; Rubin, B.K.; Voynow, J.A. Neutrophil elastase correlates with increased sphingolipid content in cystic fibrosis sputum. Pediatr. Pulmonol. 2018, 53, 872–880. [Google Scholar] [CrossRef]
- Song, L.; Wang, D.; Abbas, G.; Li, M.; Cui, M.; Wang, J.; Lin, Z.; Zhang, X.E. The main protease of SARS-CoV-2 cleaves histone deacetylases and DCP1A, attenuating the immune defense of the interferon-stimulated genes. J. Biol. Chem. 2023, 299, 102990. [Google Scholar] [CrossRef]
- Li, Z.; Fang, P.; Duan, P.; Chen, J.; Fang, L.; Xiao, S. Porcine Deltacoronavirus Infection Cleaves HDAC2 to Attenuate Its Antiviral Activity. J. Virol. 2022, 96, e0102722. [Google Scholar] [CrossRef]
- Husain, M.; Harrod, K.S. Influenza A virus-induced caspase-3 cleaves the histone deacetylase 6 in infected epithelial cells. FEBS Lett. 2009, 583, 2517–2520. [Google Scholar] [CrossRef] [PubMed]
- Galvin, H.D.; Husain, M. Influenza A virus-induced host caspase and viral PA-X antagonize the antiviral host factor, histone deacetylase 4. J. Biol. Chem. 2019, 294, 20207–20221. [Google Scholar] [CrossRef] [PubMed]
- Escaffit, F.; Vaute, O.; Chevillard-Briet, M.; Segui, B.; Takami, Y.; Nakayama, T.; Trouche, D. Cleavage and Cytoplasmic Relocalization of Histone Deacetylase 3 Are Important for Apoptosis Progression. Mol. Cell. Biol. 2007, 27, 554–567. [Google Scholar] [CrossRef][Green Version]
- Paroni, G.; Mizzau, M.; Henderson, C.; Del Sal, G.; Schneider, C.; Brancolini, C. Caspase-dependent Regulation of Histone Deacetylase 4 Nuclear-Cytoplasmic Shuttling Promotes Apoptosis. Mol. Biol. Cell 2004, 15, 2804–2818. [Google Scholar] [CrossRef]
- Liu, F.; Dowling, M.; Yang, X.-J.; Kao, G.D. Caspase-mediated Specific Cleavage of Human Histone Deacetylase 4. J. Biol. Chem. 2004, 279, 34537–34546. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xavier, S.; Moskowitz-Kassai, E.; Chen, R.; Lu, C.Y.; Sanduski, K.; Špes, A.; Turk, B.; Goligorsky, M.S. Cathepsin Cleavage of Sirtuin 1 in Endothelial Progenitor Cells Mediates Stress-Induced Premature Senescence. Am. J. Pathol. 2012, 180, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.-H.; Lee, S.-J.; Lee, K.-H.; Um, H.-D.; Kim, J.-H.; Kim, S.-J.; Kim, J.-I.; Hwang, S.-G. Ionizing Radiation Induces Cellular Senescence of Articular Chondrocytes via Negative Regulation of SIRT1 by p38 Kinase. J. Biol. Chem. 2010, 285, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Repnik, U.; Knezevic, M.; Jeras, M. Simple and cost-effective isolation of monocytes from buffy coats. J. Immunol. Methods 2003, 278, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Winkler, A.R.; Nocka, K.H.; Sulahian, T.H.; Kobzik, L.; Williams, C.M.M. In vitro modeling of human alveolar macrophage smoke exposure: Enhanced inflammation and impaired function. Exp. Lung Res. 2008, 34, 599–629. [Google Scholar] [CrossRef]
- Bell, C.W.; Jiang, W.; Reich, C.F.; Pisetsky, D.S. The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef]
- McKelvey, M.C.; Weldon, S.; McAuley, D.F.; Mall, M.A.; Taggart, C.C. Targeting Proteases in Cystic Fibrosis Lung Disease. Paradigms, Progress, and Potential. Am. J. Respir. Crit. Care Med. 2020, 201, 141–147. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Antibody (Dilution) | Target | Antibody (Dilution) |
|---|---|---|---|
| HDAC1 | sc-81598 (1:1000) | HDAC10 | sc-393417 (1:500) |
| HDAC2 | CST #5113s (1:1000) | HDAC11 | sc-390737 (1:500) |
| HDAC3 | CST #3949s (1:1000) | Sirt 1 | CST 9475T (1:1000) |
| HDAC4 | sc-46672 (1:500) | Sirt 2 | CST 12650 (1:1000) |
| HDAC5 | sc-133106 (1:500) | Sirt 3 | CST 5490 (1:1000) |
| HDAC6 | CST #7558s (1:2000) | Sirt 5 | CST 8782 (1:1000) |
| HDAC7 | sc-74563 (1:500) | Sirt 6 | CST 12486 (1:1000) |
| HDAC8 | sc-374180 (1:500) | Sirt 7 | CST 5360 (1:1000) |
| Β-actin | Sigma A5441 (1:8000) | HMGB1 | sc-56698 (1:1000) |
| Mouse IgG, HRP | NXA931 (1:10,000) | rabbit IgG, HRP | CST 7074 (1:2000) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, S.; Bulut, G.B.; Kummarapurugu, A.B.; Ma, J.; Voynow, J.A. Neutrophil Elastase Degrades Histone Deacetylases and Sirtuin 1 in Primary Human Monocyte Derived Macrophages. Int. J. Mol. Sci. 2024, 25, 4265. https://doi.org/10.3390/ijms25084265
Zheng S, Bulut GB, Kummarapurugu AB, Ma J, Voynow JA. Neutrophil Elastase Degrades Histone Deacetylases and Sirtuin 1 in Primary Human Monocyte Derived Macrophages. International Journal of Molecular Sciences. 2024; 25(8):4265. https://doi.org/10.3390/ijms25084265
Chicago/Turabian StyleZheng, Shuo, Gamze B. Bulut, Apparao B. Kummarapurugu, Jonathan Ma, and Judith A. Voynow. 2024. "Neutrophil Elastase Degrades Histone Deacetylases and Sirtuin 1 in Primary Human Monocyte Derived Macrophages" International Journal of Molecular Sciences 25, no. 8: 4265. https://doi.org/10.3390/ijms25084265
APA StyleZheng, S., Bulut, G. B., Kummarapurugu, A. B., Ma, J., & Voynow, J. A. (2024). Neutrophil Elastase Degrades Histone Deacetylases and Sirtuin 1 in Primary Human Monocyte Derived Macrophages. International Journal of Molecular Sciences, 25(8), 4265. https://doi.org/10.3390/ijms25084265