Abstract
Leber hereditary optic neuropathy (LHON) is a rare disorder causing a sudden painless loss of visual acuity in one or both eyes, affecting young males in their second to third decade of life. The molecular background of the LHON is up to 90%, genetically defined by a point mutation in mitochondrial DNA. Recently, an autosomal recessive form of LHON (LHONAR1, arLHON) has been discovered, caused by biallelic variants in the DNAJC30 gene. This study provides the results of the DNAJC30 gene analysis in a large group of 46 Polish patients diagnosed with LHON, together with the clinical characterization of the disease. The c.152A>G (p.Tyr51Cys) substitution in the DNAJC30 gene was detected in all the patients as homozygote or compound heterozygote. Moreover, we identified one novel variant, c.293A>G, p.(Tyr98Cys), as well as two ultra-rare DNAJC30 variants: c.293A>C, p.(Tyr98Ser), identified to date only in one individual affected with LHONAR1, and c.130_131delTC (p.Ser44ValfsTer8), previously described only in two patients with Leigh syndrome. The patients presented here represent the largest group of subjects with DNAJC30 gene mutations described to date. Based on our data, the autosomal recessive form of LHON caused by DNAJC30 gene mutations is more frequent than the mitochondrial form in Polish patients. The results of our study suggest that Sanger sequencing of the single-exon DNAJC30 gene should be a method of choice applied to identify a molecular background of clinically confirmed LHON in Polish patients. This approach will help to reduce the costs of molecular testing.
1. Introduction
Leber hereditary optic neuropathy (LHON, MIM: #535000; ORPHA: #104) is a rare disorder manifested by a sudden painless loss of visual acuity in one or both eyes. The disease has a strong male preponderance (80–90%) [1]. LHON mainly affects young males in their second to third decade of life, usually between ages 15 and 35, but symptoms can also be seen in childhood or at an older age [1,2]. The prevalence of the disease is about 1:30,000 in Europe. LHON displays a sex-dependent incomplete penetrance higher in men (about 50%) than in women (10%) [3,4].
LHON is a neurodegenerative disease of the optic nerve, causing the initial dysfunction of retinal ganglion cells, followed by axon degeneration. The first symptoms of the disease include a sudden acute or subacute painless, unilateral loss of central vision with involvement of the other eye after a few weeks to months later or affecting both eyes simultaneously. Visual acuity loss generally stabilizes within 4–6 months, and in most patients, the visual acuity is worse than 20/200. Visual field defects are typically central or cecocentral. Most patients observe central scotoma, usually at the early stage of the disease, but later, visual field defects become larger. Among the symptoms of LHON, there is also the early impairment of color perception [1,5].
During the first 4–5 weeks of the disease, which is called the acute phase, the eye fundus and OCT examinations usually reveal an elevation of the optic nerve disc and perivascular telangiectatic microangiopathy, as well as the swelling of the retinal nerve fiber layer, which, unfortunately, is often mistakenly considered a sign of an inflammatory process. In approximately 20% of patients, the eye fundus may look normal during the acute phase of LHON, which can delay the accurate diagnosis [6]. Eventually, the signs of the chronic phase become apparent after a few months. Funduscopy and OCT reveal optic disc pallor, thinning, and the atrophy of the retinal nerve fiber layer and ganglion cell complex at this phase of LHON. The disease progresses rapidly, with only a negligible probability of visual recovery depending on the type of mitochondrial mutation [2,4]. However, in more than 45% of patients, visual acuity improved after being treated with idebenone, a short-chain synthetic ubiquinone analog [7,8].
Leber hereditary optic neuropathy is known to be the most frequent mitochondrial disease. The molecular background of the LHON is up to 90%, genetically defined by a point mutation in mitochondrial DNA. The vast majority of mitochondrial LHON (mtLHON) cases are caused by one of three primary mitochondrial mutations within the genes encoding mitochondrial complex I subunits (NADH–ubiquinone oxidoreductase): m.11778G>A (localized in the MT-ND4 gene), m.3460G>A (within the MT-ND1 gene), and m.14484T>C (within the MT-ND6 gene) [2,4].
Recently, an autosomal recessive form of Leber hereditary optic neuropathy (LHONAR1, arLHON, MIM: #619382) has been discovered. The autosomal recessive form of LHON is mainly caused by biallelic variants in the DNAJC30 (MIM: 618202) gene [8,9]. Typical LHON phenotypes have also been reported to be the effects of variants in other genes, namely NDUFS2, MCAT, and NDUFA12, detected only in a few families [10,11,12,13]. Pathogenic variants in the DNAJC30 gene were reported to affect the mitochondrial complex I subunit and interact with the mitochondrial adenosine triphosphate (ATP) synthesis [9].
The DNAJC30 encoding the DNAJ/HSP40 homolog, subfamily C, member 30 protein, controls ATP synthase activity and plays a leading role in the repair mechanism of oxidative phosphorylation complex I. DNAJC30 is a chaperone protein expressed mainly in neurons. It is required to efficiently exchange complex I subunits exposed to reactive oxygen species and integral to a mitochondrial complex I repair mechanism [9]. The affected DNAJC30 protein is incapable of regenerating the damaged complex I subunits. It results in low ATP synthesis and high levels of reactive oxygen species, which causes the degeneration of retinal ganglion cells. Similarly to mtLHON, the autosomal recessive form of LHON also demonstrates sex-dependent incomplete penetrance with more affected men than women [9].
About 100 LHON patients with DNAJC30 mutations have been reported [8,9,10]. Approximately 85% of patients originate from Eastern Europe (Russia, Ukraine, Poland, and Romania). Most patients were affected with the c.152A>G variant, causing the amino acid substitution of tyrosine at position 51 within the conservative domain of DNAJC30 protein to cysteine. The p.Tyr51Cys substitution is a founder mutation in Eastern Europe [9].
From the year 2008 until April 2021, the routinely performed genetic testing in Polish patients with suspected Leber hereditary optic neuropathy was basically restricted to Sanger sequencing of three mtDNA regions encompassing three primary LHON-causing mutations: m.11778G>A, m.3460G>A, and m.14484T>C. Some patients with no mtDNA mutations detected based on this diagnostic strategy had whole mitochondrial genome sequencing performed. Moreover, the DNA sequencing of the genes associated with optic atrophies was performed in some individuals.
In April 2021, following the report on the DNAJC30 gene variants as a cause of the autosomal recessive form of LHON [9], we implemented molecular testing for DNAJC30 variants in those patients in whom no changes in the mtDNA were detected. Although it is known that mtDNA mutations cause most LHON cases, many Polish patients with the typical course of the disease have been waiting for a molecular diagnosis for many years until we implemented the DNAJC30 gene sequencing.
This study aimed to report a clinical characterization and molecular basis of the autosomal recessive form of Leber hereditary optic neuropathy in 46 Polish patients, the largest cohort of patients with the LHONAR1 reported to date. Based on our data, the autosomal recessive LHON caused by DNAJC30 gene mutations is more frequent than the mitochondrial form in Polish patients. Moreover, by reporting a novel variant, this study broadens the mutation spectrum of probable pathogenic DNAJC30 variants. In this paper, we also show the diagnostic odyssey undergone by some of our patients, which is no longer needed since a simple, inexpensive molecular test for variants’ screening in one small gene enables establishing a molecular diagnosis in most Polish LHON patients.
2. Results
From April 2021, all the patients with no mtDNA variants detected and those referred to our clinic for the first time were offered molecular testing for DNAJC30 gene variants. Here, we present the results of the DNAJC30 gene analysis in a large group of 46 Polish patients clinically diagnosed with Leber hereditary optic neuropathy.
2.1. Clinical Diagnosis of Leber Hereditary Optic Neuropathy
Most patients’ first symptom of LHON was the deterioration or loss of visual acuity. It mostly involved one eye first, followed by the other eye over three days to two years (usually, the fellow eye was affected after two weeks to three months). The average age of LHON onset among our patients was 20.5 years (19.5 years, excluding patient no. 40_45, whose age of onset was 68). The average age of onset among affected women was the same as that in a group of men (when excluding patient no. 40_45) and was equal to 19 years. Fundus examination showed hyperemic optic nerves during the acute phase of LHON in most patients. Moreover, peripapillary microangiopathy was observed at the beginning of the disease in many patients. Most patients reported centrocecal scotoma or other visual field defects, and some patients noticed impaired color perception. Table 1 presents the clinical symptoms observed in the 46 patients.

Table 1.
Clinical symptoms in 46 Polish patients affected with the DNAJC30 variants.
2.2. Molecular Genetic Analysis
The sequencing analysis of the three mtDNA regions encompassing three primary LHON-causing mutations performed in one proband from each family (40 subjects), excluding patient no. 23_26 from family 23, did not reveal any causative mitochondrial DNA variants. Twenty-two patients (patients’ ID numbers are listed in Materials and Methods) with no variants found in the three analyzed mtDNA fragments were subjected to whole mitochondrial genome sequencing, which did not lead to the detection of any potentially pathogenic variants. Three benign variants in mitochondrial DNA were identified in three patients: mt.11253T>C (in patient no. 3_3), mt.14199T>C (in the patient no. 8_9), and mt.11654A>G (in patient no. 16_18).
Before the DNAJC30 gene screening was implemented in diagnostics, some patients from our study group underwent additional molecular tests. Multiplex ligation-dependent probe amplification (MLPA), performed in patients no. 15_17, 18_20, and 21_24, failed to identify the molecular background of the disease. Single-nucleotide polymorphism (SNP) microarray based on APEX technology for detecting variants in the OPA1 gene carried out in patients no. 15_17 and 18_20 also did not reveal any causative variants. Moreover, a next-generation sequencing (NGS) panel encompassing genes associated with optic atrophy (excluding the DNAJC30 gene at that time) and an NGS panel for genetically determined retinal diseases encompassing 274 genes performed in patient no. 15_17 did not detect any potentially pathogenic variants.
Patients with the still unknown molecular basis of LHON were subjected to Sanger sequencing of the DNAJC30 gene (all patients excluding patients no. 20_22, 23_26, and 40_45) or the NGS panel encompassing the genes associated with optic nerve atrophy that included this gene (patients no. 20_22, 23_26, and 40_45). The recurrent variant c.152A>G (p.Tyr51Cys) was identified in all the examined patients, either in the homozygous state (in 41 patients) or in the form of compound heterozygote along with another variant on a second allele (in 5 patients).
We discovered a novel variant, c.293A>G, p.(Tyr98Cys), in patient no. 35_40. The results of the segregation analysis in the family were consistent with the autosomal recessive mode of inheritance (see the pedigree and chromatogram in Figure 1).
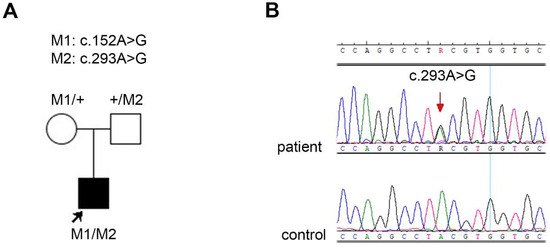
Figure 1.
(A) Pedigree of the family 35 together with the segregation analysis results. The proband (patient no. 35_40) is marked with an arrow and a square filled with black. Unfilled symbols indicate unaffected, heterozygous parents. (B) Chromatogram showing the novel variant c.293A>G (indicated with the red arrow) in patient no. 35_40.
The variant was not reported in the following databases Leiden Open Variation Database (LOVD, http://www.lovd.nl/3.0/home, accessed on 10 May 2023, Varsome (Varsome The Human Genomics Community, https://varsome.com/, accessed on 10 May 2023), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar, accessed on 10 May 2023), and GnomAD Browser (Genome Aggregation Database, https://gnomad.broadinstitute.org/, accessed on 10 May 2023). According to the American College of Medical Genetics and Genomics (ACMG) classification guidelines, the variant is predicted to be pathogenic. The nucleotide adenine at position 293 and the codon TAC are conserved between species (Figure 2A). The p.(Tyr98Cys) variant is located within the conserved region of the protein, the J domain, which is crucial for the functional interactions of the DNAJC30 protein. Moreover, tyrosine at the amino acid position 98 is highly conserved between species (Figure 2B). The in silico predictions of the variant’s potential pathogenicity with the use of SIFT (score: <0.05, deleterious) and PolyPhen-2 (score: 1, probably damaging) indicated that the substitution is damaging.
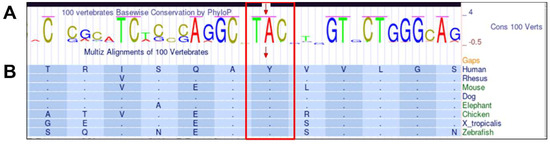
Figure 2.
The conservation of the novel variant c.293A>G, p.(Tyr98Cys) based on the UCSC Genome Browser. The upper panel, (A), shows PhyloP results presenting the conservation of adenine and TAC codon in 100 vertebrates, and the lower panel, (B), shows Multiz alignments of amino acid tyrosine at position 98 conservation between selected vertebrate species.
Interestingly, we identified a different substitution at the same nucleotide position, an ultra-rare variant c.293A>C, p.(Tyr98Ser). We detected this variant in a compound heterozygous state in two brothers (patients no. 28_31 and 28_32). According to ACMG classification guidelines, the variant is predicted to be likely pathogenic. The in silico predictions of the p.(Tyr98Ser) potential pathogenicity using SIFT indicated that the substitution affects protein function (score: 0, deleterious). Our analysis with PolyPhen-2 showed that p.(Tyr98Ser) is probably damaging (score: 0.99). To the best of our knowledge, the substitution c.293A>C was only reported once in the ClinVar database in one patient affected with autosomal recessive Leber hereditary optic neuropathy (VCV001299584.1). The variant was identified in the GnomAD Browser as a heterozygote in 1 out of the 249,566 analyzed alleles in healthy individuals.
Moreover, in two patients, we identified an extremely rare deletion in the DNAJC30 gene: c.130_131delTC (p.Ser44ValfsTer8). In both affected men, no. 1_1 and no. 22_25, the deletion was identified in a compound heterozygote state with c.152A>G (p.Tyr51Cys). This frameshift variant was classified as likely pathogenic. The substitution was previously identified in patients with Leigh syndrome: in one Polish family [14] and a Moroccan boy [15].
All DNAJC30 gene variants identified in our patients are listed in Table 2.
Table 2.
DNAJC30 gene variants identified in the group of 46 Polish patients.
3. Discussion
Here, we report the DNAJC30 gene variants in 46 Polish patients with a clinical diagnosis of LHON. This is the first report on DNAJC30 variants in such a significant cohort of patients, especially since they come from one country. To date, the autosomal recessive form of LHON has been diagnosed in about one hundred patients, most of whom were affected with DNAJC30 gene variants [10].
Ophthalmological findings in our group of patients with autosomal recessive LHON were indistinguishable from those of the mitochondrial LHON form. The disease course was typical for LHON, usually involving acute painless central vision loss and chronic phase. No patients in our study group presented extraocular symptoms of LHON (LHON plus).
The average age of disease onset was 19 years when assessing both sexes together, as well as when assessing men and women separately. It is consistent with the already published data for men but differs from the reported data for women. Leaners and coworkers reported that the age of the autosomal recessive form of LHON onset in 10 affected women was higher than in our female patients and amounted to 32 years [10]. In our female patients, the disease began earlier, as the youngest patient was six years old at the age of LHONAR1 onset, and the oldest was 29. Interestingly, in this study, we also present a patient with very late disease onset, patient no. 40_45, who had first symptoms at the age of 68 years. The appearance of the first symptoms of Leber hereditary optic neuropathy in patients over 50 years of age is rare. However, cases of the mitochondrial LHON form with late disease onset have been reported [16,17,18]. It is worth noting that, to our knowledge, patient no. 40_45 is the first to be reported with an autosomal recessive form of LHON with such a late onset of the disease.
Diagnostic difficulties concerned not only this patient but also those patients in our study in whom, two or more years ago, no mitochondrial variants were identified. Over all these years, before the autosomal recessive form of LHON was reported, many patients with the typical course of LHON remained undiagnosed. These patients underwent a veritable diagnostic odyssey, waiting for a molecular diagnosis for even ten years. Some of them have had a large number of molecular tests performed, e.g., patient no. 15_17, who underwent MLPA and SNP microarray based on APEX technology for the OPA1 gene as well as the NGS panel encompassing the genes associated with autosomal dominant optic atrophy and the NGS panel for genetically determined retinal diseases (without the DNAJC30 gene included at that time). Moreover, the clinical diagnosis of LHON is complicated, especially if the patient is examined for the first time in the chronic phase of the disease when the clinical picture of the disorder resembles optic nerve atrophy caused by many other factors.
The other group of patients in the presented cohort are those affected with LHON who came to the genetic clinic within the last two years. Since the DNAJC30-related LHONAR was reported, the molecular diagnostic approach in patients with clinical features of LHON is relatively uniform in Polish patients. The most common procedure involves searching for the three most common variants in mtDNA or the whole mitochondrial genome sequencing and Sanger sequencing of the single-exon DNAJC30 gene coding region. Nevertheless, three of the described patients had the NGS panel for genetically determined retinal diseases (encompassing 274 genes, including the DNAJC30 gene), which was performed instead of the DNAJC30 gene sequencing due to the suggestion of different optic nerve atrophy clinical diagnosis or due to the patient’s will. However, Sanger sequencing of the DNAJC30 gene coding sequence is the low-cost, fast, and effective method in searching for the molecular background of LHON in Polish patients, suggesting that it should be implemented as the first-step test.
From 2008 to April 2023, we diagnosed the mitochondrial form of LHON in 102 patients examined in one of the genetic centers where molecular tests for LHON reported in this study were carried out. On the other hand, since we introduced the DNAJC30 gene test two years ago, we diagnosed LHONAR1 in 32 patients (out of a described group of 46) in this genetic center. Comparing these data, we can conclude that the autosomal recessive form of LHON is much more frequent in Polish patients than the classic mitochondrial form. The relatively high prevalence of DNAJC30 variants in patients with LHON supports the thesis that the DNAJC30 gene analysis should be the test of choice in Polish patients with LHON.
The rapid molecular diagnosis of patients with LHON is crucial to undertake fast treatment, which is necessary to stop the process of optic nerve degeneration. Unfortunately, we have scarce data on the number of patients who applied Idebenon treatment and the effects of treatment in our group of patients. This results from the fact that many patients do not make follow-up appointments in the genetic clinic, and some do not even have a routine ophthalmic examination once their vision is stabilized.
The c.152A>G (p.Tyr51Cys) substitution is known to be the most common variant in the DNAJC30 gene and a founder mutation. This substitution accounts for over 20% of molecularly diagnosed patients in Russia [9] and 90% of disease-associated variants in the German cohort. Interestingly, our research shows that the p.Tyr51Cys variant is widespread in Polish patients, as it was identified in all subjects with a frequency of nearly 95% of alleles. Moreover, we identified a novel variant c.293A>G, p.(Tyr98Cys) within the conserved region of the protein crucial for the DNAJC30 protein interactions. Functional analyses are required to explain the impact of the novel variant and two other variants detected in our patients, p.(Tyr98Ser) and p.(Ser44ValfsTer8), on the DNAJC30 protein function. No genotype–phenotype correlations were found in our cohort of patients.
4. Material and Methods
4.1. Patients
This report focuses on 46 patients from 41 Polish families with clinical symptoms of Leber hereditary optic neuropathy, in whom no mtDNA variants were identified. Still, the DNAJC30 gene screening fully confirmed the clinical diagnosis. There were 41 affected men and 5 women in our study group. The age of disease onset in this group of patients with LHON was 6 to 68 years old.
The patients were numbered with Patient ID, where the first digit indicated the family number, and the next digit after the underscore character was the individual’s laboratory number. This study was conducted by the tenets of the Declaration of Helsinki and the Association for Research in Vision and Ophthalmology (ARVO) statement on human subjects. Written informed consent was obtained from all participants or their parents or legal guardians if patients were under 16 years old.
4.2. Molecular Analysis
4.2.1. Mitochondrial DNA Analyses
The sequencing analysis of three mtDNA regions encompassing three primary LHON-causing mutations, namely m.11778G>A, m.3460G>A, and m.14484T>C, was performed in one proband from each family (40 subjects), excluding family 23 (patient no. 23_26). The primers used for amplification and sequencing, and the polymerase chain reaction (PCR) conditions are available upon request.
Until April 2021, the whole mitochondrial genome sequencing was performed in 22 patients, in whom no variants were detected in the previously analyzed mtDNA regions based on Sanger sequencing (patients no. 1_1; 2_2; 3_3; 14_16; 15_17; 16_18, 17_19, 18_20, 19_21, 20_22, 22_25, 24_27, 25_28, 26_29, 27_30, 28_31, 29_33, 30_34, 31_35, 32_37, 33_38, and 34_39). Mitochondrial DNA for next-generation sequencing was amplified in one fragment encompassing the whole molecule or, in case of lower DNA quality, in two overlapping fragments. Genomic libraries were created using the Nextera XT DNA Sample Preparation Kit and Nextera XT Index Kit (Illumina Inc., Foster City, CA, USA) and then sequenced using NGS technology on the MiSeq platform and the MiSeq Reagent Kit v3 (Illumina). The obtained data were subjected to bioinformatics analysis, aligned to the reference mtDNA sequence, rCRS, and subjected to variant identification using the CLC Genomics Workbench (CLC bio) software (v11). Variant pathogenicity was assessed using the MITOMAP database (https://www.mitomap.org/MITOMAP, accessed on 1 March 2021). Patients who came to the genetic clinic after April 2021 did not have whole mitochondrial genome sequencing performed. For details on the whole mitochondrial genome sequencing procedure, see Piotrowska-Nowak et al., 2019 [19].
4.2.2. Molecular Analyses of Nuclear Genes Associated with Optic Nerve Atrophies (Excluding DNAJC30)
The NGS panel for the detection of variants in three genes (OPA1, OPA3, and TMEM126A) associated with autosomal dominant optic atrophy (ADOA) was performed (Genomed, Warsaw, Poland) in two patients: no. 15_17 and 21_24.
Multiplex ligation-dependent probe amplification (MLPA)-based assay for the detection of OPA1 gene deletions and duplications (Salsa MLPA Probemix, P229 OPA1, MRC-Holland, Amsterdam, The Netherlands) was carried out in three patients: no. 15_17, 18_20, and 21_24.
SNP microarray-based on APEX technology, for detecting 122 SNPs (version 1.0) in the OPA1 gene (Asper Biotech Ltd., Tartu, Estonia), was applied in patients no. 15_17 and 18_20. Moreover, patient no. 15_17 also underwent the NGS panel for genetically determined retinal diseases encompassing 274 genes (Genomed, Warsaw, Poland). Still, the panel version at that time did not include the DNAJC30 gene (see Supplementary Materials S1a: List of 274 genes analyzed on NGS retinal diagnostic panel).
4.2.3. DNAJC30 Gene Analyses
The fragment encompassing the coding sequence of the single-exon gene DNAJC30 was amplified via PCR and sequenced with the Sanger sequencing method in 43 patients (all of them excluding patients no. 20_22, 23_26, and 40_45). The DNAJC30 gene screening was performed at two genetic centers using two primer pairs designed based on the NM_032317.3 reference sequence. The PCR using the following primers F: 5′gtttCTCTTGCACCGCCTG3′ and 5′GACCGATACTCCTGCCGTTT3′ was performed in 25 patients. The primers F: 5′CTTTACGTGACTGGCCACAG3′ and R: 5′CTATCAATGGCCAAGGGTTC3′ elongated with a universal M13 sequence tag at the 5′ ends (forward 5′TGTAAAACGACGGCCAGT3′ and reverse 5′CAGGAAACAGCTATGACC3′) were applied for the molecular testing of 20 patients. The polymerase chain reaction (PCR) conditions are available upon request. The PCR products were purified with the ExoSAP-IT kit (Exonuclease I and Shrimp Alkaline Phosphatase Cleanup for PCR products, Affymetrix, Santa Clara, CA, USA) and bidirectly sequenced using dye-terminator chemistry (v3.1BigDye® Terminator Thermo Fisher Scientific, Waltham, MA, USA). The sequencing products were separated on an ABI 3130xl capillary sequencer (Applied Biosystems, Thermo Fisher Scientific).
The NGS panel for the detection of variants in 26 or 30 genes associated with optic nerve atrophy pathogenesis (two versions, both including the DNAJC30 gene) (Genomed, Warsaw, Poland) was performed in three patients: no. 20_22, 23_26, and 40_45 (see Supplementary Material S1b: List of genes analyzed on NGS panel for optic nerve atrophy (OKUA-NGS)).
The novel DNAJC30 gene substitution, as well as two ultra-rare variants, were cross-checked with the following databases Leiden Open Variation Database (LOVD), Varsome (Varsome The Human genomics community), ClinVar, and GnomAD Browser (Genome Aggregation Database). We annotated the novel variant against the DNAJC30 gene reference sequence NM_032317.3, following the nomenclature guidelines of the Human Genome Variation Society (HGVS, http://varnomen.hgvs.org/, accessed on 10 May 2023). The pathogenicity of the identified variants was assessed according to the American College of Medical Genetics and Genomics (ACMG) classification [20].
In silico analysis was performed to predict the possible effects of the identified novel or rare missense variants on protein function. The following tools were used: SIFT (Sorting Intolerant from Tolerant, https://sift.bii.astar.edu.sg/), PROVEAN (Protein Variation Effect Analyzer, http://provean.jcvi.org/), and PolyPhen-2 (Polymorphism Phenotyping v.2, http://genetics.bwh.harvard.edu/pph2/) (accessed on 10 July 2023).
5. Conclusions
To summarize, our group of patients affected with DNAJC30 gene variants, encompassing 46 patients of Polish origin, is the largest cohort of patients with LHONAR1 described to date. Based on our data, the autosomal recessive form of LHON is more frequent than the mitochondrial form in Polish patients. We determined the substitution c.152A>G as the most common variant in the DNAJC30 gene among Polish patients. Moreover, through the identification of a novel variant c.293A>G, p.(Tyr98Cys), this study broadens the mutation spectrum of probable pathogenic DNAJC30 variants.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms242417496/s1.
Author Contributions
Conceptualization, A.S.-W., K.T. and M.R.K.; methodology, A.S.-W. and K.T.; validation, A.W. and K.T.; formal analysis, K.T.; investigation, A.S.-W., K.T. and A.M.; resources, A.S.-W., K.N., M.K., M.O. and M.R.K.; data curation, A.S.-W. and K.T.; writing—original draft preparation, A.S.-W.; writing—review and editing, A.S.-W. and K.T.; visualization, A.S.-W.; supervision, M.R.K.; project administration, K.T.; funding acquisition, K.T. All authors have read and agreed to the published version of the manuscript.
Funding
The study was partially supported by grant no. BOB-IDUB-622/179/2021 to K.T.
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki and was based on the retrospective analysis of the diagnostic results. According to Polish law, it is not a medical experiment and does not need any opinion of the Bioethical Committee.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data used in this study is available from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shemesh, A.; Sood, G.; Margolin, E. Leber Hereditary Optic Neuropathy (LHON); StatPearls: St. Petersburg, FL, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482499/ (accessed on 2 October 2023).
- Man, P.Y.W.; Turnbull, D.M. Leber Hereditary Optic Neuropathy. J. Med. Genet. 2002, 39, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Votruba, M.; Burté, F.; La Morgia, C.; Barboni, P.; Carelli, V. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. 2016, 132, 789–806. [Google Scholar] [CrossRef]
- Sundaramurthy, S.; SelvaKumar, A.; Ching, J.; Dharani, V.; Sarangapani, S.; Yu-Wai-Man, P. Leber hereditary optic neuropathy-new insights and old challenges. Graefes Arch. Clin. Exp. Ophthalmol. 2021, 259, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Votruba, M.; Moore, A.T.; Chinnery, P.F. Treatment strategies for inherited optic neuropathies: Past, present and future. Eye (Basingstoke) 2014, 28, 521–537. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Chinnery, P.F. Leber Hereditary Optic Neuropathy. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2000; pp. 1993–2023. [Google Scholar]
- Catarino, C.B.; Von Livonius, B.; Priglinger, C.; Banik, R.; Matloob, S.; Tamhankar, M.A.; Castillo, L.; Friedburg, C.; Halfpenny, C.A.; Lincoln, J.A.; et al. Real-World Clinical Experience with Idebenone in the Treatment of Leber Hereditary Optic Neuropathy. J. Neuro-Ophthalmol. 2020, 40, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Tesarova, M.; Sheremet, N.L.; Catarino, C.B.; Carelli, V.; Ciara, E.; Curry, K.; Engvall, M.; Fleming, L.R.; Freisinger, P.; et al. DNAJC30 defect: A frequent cause of recessive Leber hereditary optic neuropathy and Leigh syndrome. Brain 2022, 145, 1624–1631. [Google Scholar] [CrossRef]
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Investig. 2021, 131, e138267. [Google Scholar] [CrossRef]
- Lenaers, G.; Beaulieu, C.; Charif, M.; Gerber, S.; Kaplan, J.; Rozet, M. Autosomal recessive Leber hereditary optic neuropathy, a new neuro-ophthalmo-genetic paradigm. Brain 2023, 146, 3156–3161. [Google Scholar] [CrossRef] [PubMed]
- Magrinelli, F.; Cali, E.; Braga, V.L.; Yis, U.; Tomoum, H.; Shamseldin, H.; Raiman, J.; Kernstock, C.; Rezende Filho, F.M.; Barsottini, O.G.P.; et al. Biallelic Loss-of-Function NDUFA12 Variants Cause a Wide Phenotypic Spectrum from Leigh/Leigh-Like Syndrome to Isolated Optic Atrophy. Mov. Disord. Clin. Pract. 2022, 9, 218–228. [Google Scholar] [CrossRef]
- Gerber, S.; Ding, M.G.; Gérard, X.; Zwicker, K.; Zanlonghi, X.; Rio, M.; Serre, V.; Hanein, S.; Munnich, A.; Rotig, A.; et al. Compound heterozygosity for severe and hypomorphic NDUFS2 mutations cause non-syndromic LHON-like optic neuropathy. J. Med. Genet. 2017, 54, 346–356. [Google Scholar] [CrossRef]
- Gerber, S.; Orssaud, C.; Kaplan, J.; Johansson, C.; Rozet, J.M. Mcat mutations cause nuclear lhon-like optic neuropathy. Genes 2021, 12, 521. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, M.; Krygier, M.; Pawłowicz, M.; Wilke, M.V.M.B.; Rutkowska, K.; Gueguen, N.; Desquiret-Dumas, V.; Klee, E.W.; Schimmenti, L.A.; Sławek, J.; et al. Expanding the phenotype of DNAJC30-associated Leigh syndrome. Clin. Genet. 2022, 102, 438–443. [Google Scholar] [CrossRef]
- Nesti, C.; Ticci, C.; Rubegni, A.; Doccini, S.; Scaturro, G.; Vetro, A.; Guerrini, R.; Santorelli, F.M.; Procopio, E. Additive effect of DNAJC30 and NDUFA9 mutations causing Leigh syndrome. J. Neurol. 2023, 270, 3266–3269. [Google Scholar] [CrossRef] [PubMed]
- Zaslavsky, K.; Margolin, E.A. Leber’s Hereditary Optic Neuropathy in Older Individuals Because of Increased Alcohol Consumption During the COVID-19 Pandemic. J. Neuroophthalmol. 2021, 41, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, K.; Leonhardt, M.; Yu-Wai-Man, P.; Kirkman, M.A.; Korsten, A.; De Coo, I.F.; Chinnery, P.F.; Klopstock, T. Leber’s hereditary optic neuropathy with late disease onset: Clinical and molecular characteristics of 20 patients. Orphanet J. Rare Dis. 2014, 9, 158. [Google Scholar] [CrossRef]
- Pfeiffer, M.L.; Hashemi, N.; Foroozan, R.; Lee, A.G. Late-onset Leber hereditary optic neuropathy. Clin. Exp. Ophthalmol. 2013, 41, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska-Nowak, A.; Kosior-Jarecka, E.; Schab, A.; Wrobel-Dudzinska, D.; Bartnik, E.; Zarnowski, T.; Tonska, K. Investigation of whole mitochondrial genome variation in normal tension glaucoma. Exp. Eye Res. 2019, 178, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).