
The Dynamics of OXA-23 β-Lactamase from Acinetobacter baumannii




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
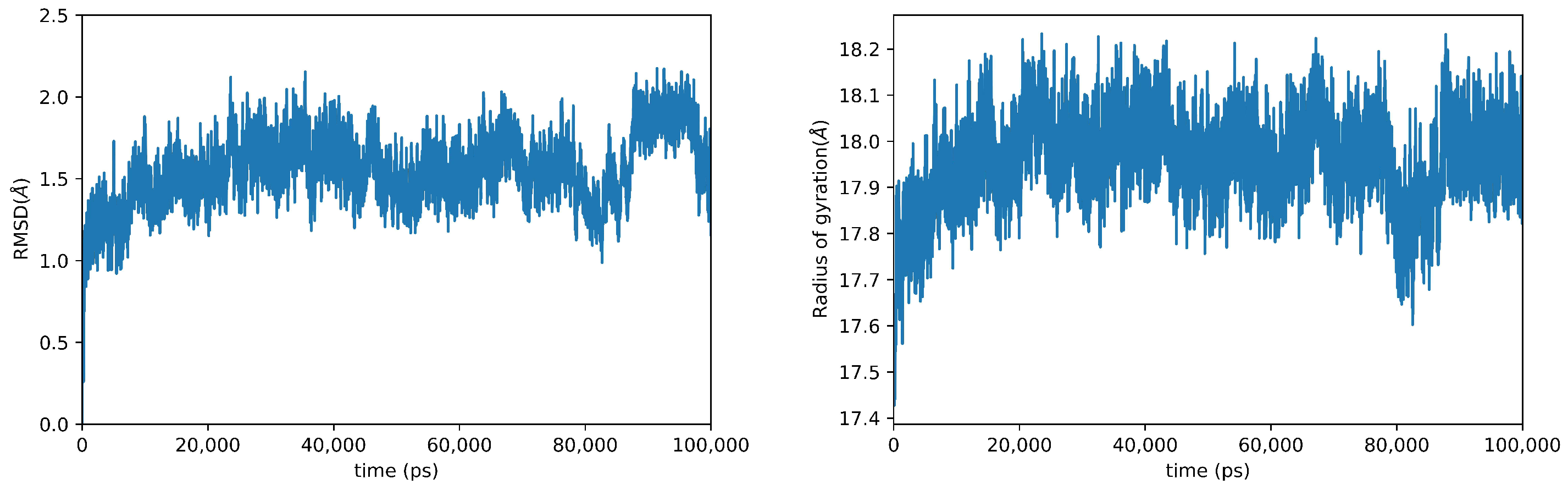
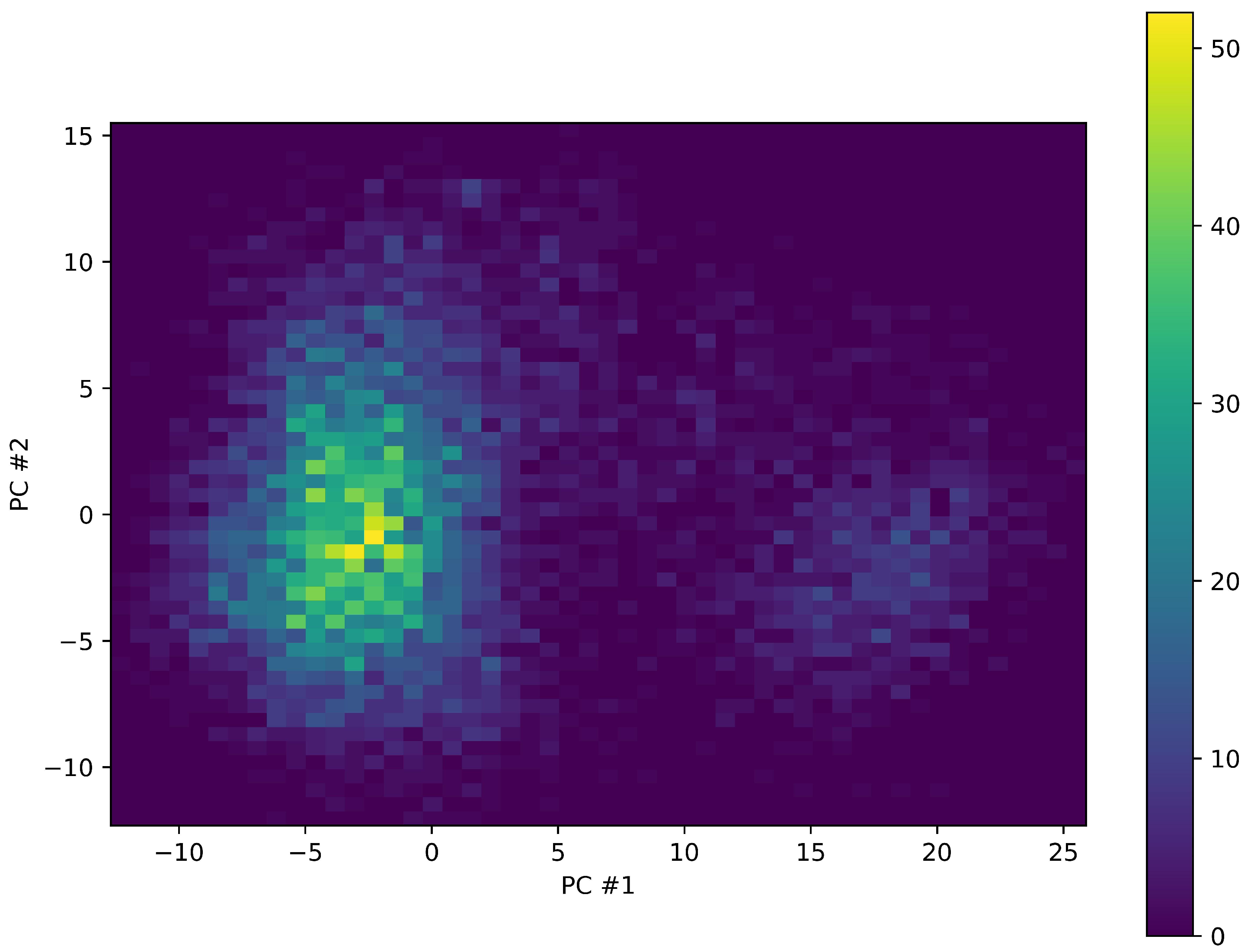
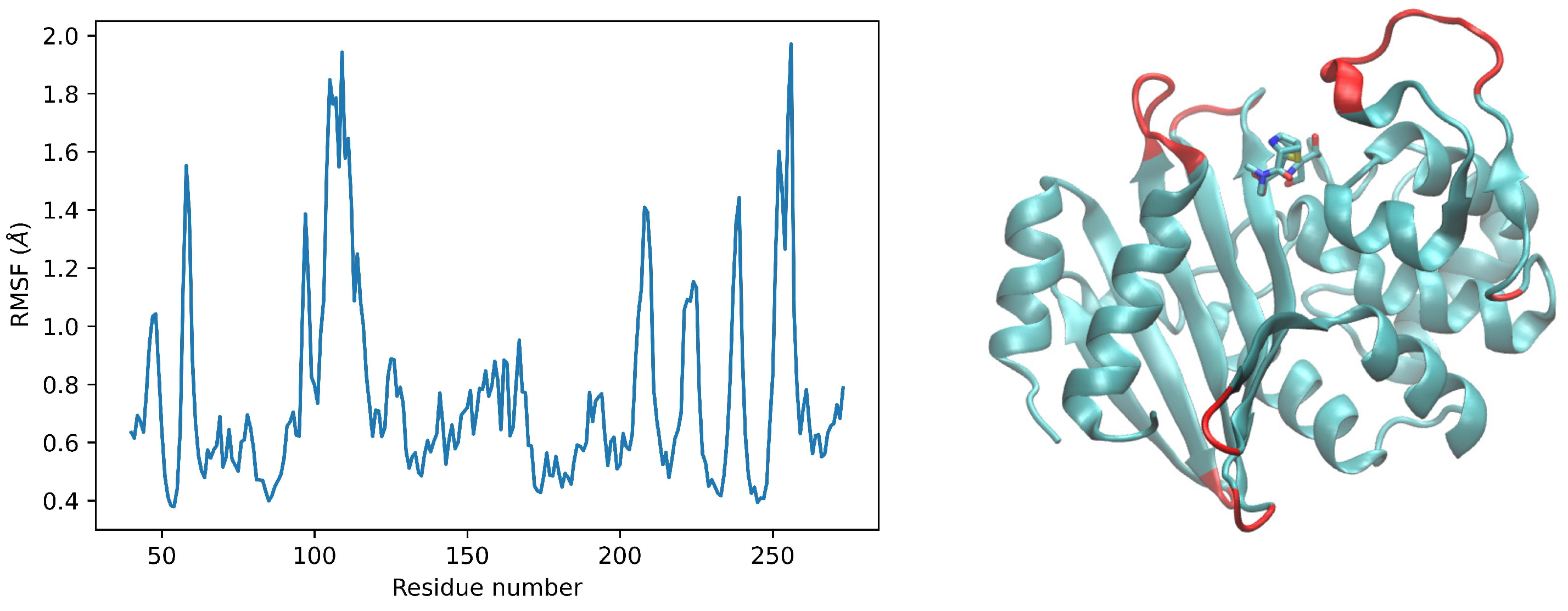
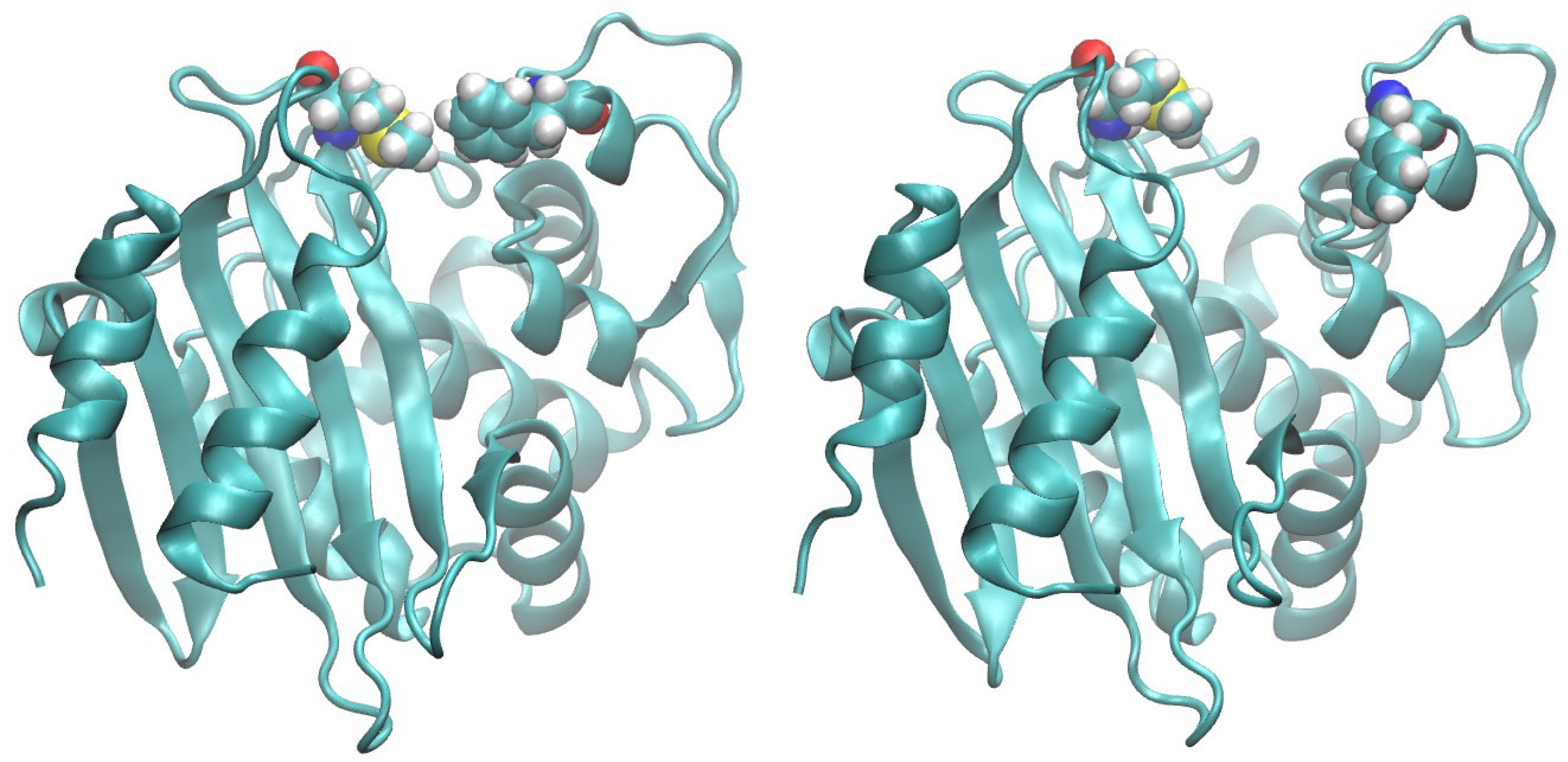
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 1929, 10, 226. [Google Scholar] [CrossRef]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, D.M.; Forde, B.M.; Kidd, T.J.; Harris, P.N.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial antibiotic resistance: The most critical pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, F.; Stolfa, S.; Morea, A.; Ronga, L.; Prete, R.D.; Chironna, M.; Santacroce, L.; Mosca, A. Meropenem/vaborbactam activity in vitro: A new option for Klebsiella pneumoniae carbapenemase (KPC)-producing Klebsiella pneumoniae treatment. Future Microbiol. 2021, 16, 1261–1266. [Google Scholar] [CrossRef]
- Arrigoni, R.; Ballini, A.; Topi, S.; Bottalico, L.; Jirillo, E.; Santacroce, L. Antibiotic Resistance to Mycobacterium tuberculosis and Potential Use of Natural and Biological Products as Alternative Anti-Mycobacterial Agents. Antibiotics 2022, 11, 1431. [Google Scholar] [CrossRef]
- Cazanave, C.; Manhart, L.E.; Bébéar, C. Mycoplasma genitalium, an emerging sexually transmitted pathogen. Med. Mal. Infect. 2012, 42, 381–392. [Google Scholar] [CrossRef]
- Hoffman, S.J.; Caleo, G.M.; Daulaire, N.; Elbe, S.; Matsoso, P.; Mossialos, E.; Rizvi, Z.; Røttingen, J.A. Strategies for achieving global collective action on antimicrobial resistance. Bull. World Health Organ. 2015, 93, 867–876. [Google Scholar] [CrossRef]
- Collier, P.; O’Neill, L.J. Two years on: An update on achievement towards the recommendations of the antimicrobial resistance report. J. Appl. Microbiol. 2018, 125, 308–312. [Google Scholar] [CrossRef]
- Davies, S.C.; Fowler, T.; Watson, J.; Livermore, D.M.; Walker, D. Annual Report of the Chief Medical Officer: Infection and the rise of antimicrobial resistance. Lancet 2013, 381, 1606–1609. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Seifert, H.; Paterson, D.L. Acinetobacter baumannii: Emergence of a successful pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef] [PubMed]
- Visca, P.; Seifert, H.; Towner, K.J. Acinetobacter infection–an emerging threat to human health. IUBMB Life 2011, 63, 1048–1054. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Bonomo, R.A.; Tolmasky, M.E. Carbapenemases: Transforming Acinetobacter baumannii into a yet more dangerous menace. Biomolecules 2020, 10, 720. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guidelines for the Prevention and Control of Carbapenem-Resistant Enterobacteriaceae, Acinetobacter baumannii and Pseudomonas aeruginosa in Health Care Facilities; World Health Organization: Geneva, Switzerland, 2017.
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Dever, L.A.; Dermody, T.S. Mechanisms of bacterial resistance to antibiotics. Arch. Intern. Med. 1991, 151, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Past and present perspectives on β-lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. Nature 1940, 146, 837. [Google Scholar] [CrossRef]
- Kirby, W.M. Extraction of a highly potent penicillin inactivator from penicillin resistant staphylococci. Science 1944, 99, 452–453. [Google Scholar] [CrossRef]
- Beta Lactamase Data Base. Available online: http://www.bldb.eu/ (accessed on 22 May 2023).
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233. [Google Scholar] [CrossRef]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1980, 289, 321–331. [Google Scholar] [PubMed]
- Jeon, J.H.; Lee, J.H.; Lee, J.J.; Park, K.S.; Karim, A.M.; Lee, C.R.; Jeong, B.C.; Lee, S.H. Structural basis for carbapenem-hydrolyzing mechanisms of carbapenemases conferring antibiotic resistance. Int. J. Mol. Sci. 2015, 16, 9654–9692. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.J.; Jeong, S.H. Class D β-lactamases. J. Antimicrob. Chemother. 2021, 76, 836–864. [Google Scholar] [CrossRef] [PubMed]
- Kaitany, K.C.J.; Klinger, N.V.; June, C.M.; Ramey, M.E.; Bonomo, R.A.; Powers, R.A.; Leonard, D.A. Structures of the class D carbapenemases OXA-23 and OXA-146: Mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins, and aztreonam. Antimicrob. Agents Chemother. 2013, 57, 4848–4855. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Antunes, N.T.; Stewart, N.K.; Toth, M.; Kumarasiri, M.; Chang, M.; Mobashery, S.; Vakulenko, S.B. Structural basis for carbapenemase activity of the OXA-23 β-lactamase from Acinetobacter baumannii. Chem. Biol. 2013, 20, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.M.; Clasman, J.R.; June, C.M.; Kaitany, K.C.J.; LaFleur, J.R.; Taracila, M.A.; Klinger, N.V.; Bonomo, R.A.; Wymore, T.; Szarecka, A.; et al. Structural basis of activity against aztreonam and extended spectrum cephalosporins for two carbapenem-hydrolyzing class D β-lactamases from Acinetobacter baumannii. Biochemistry 2015, 54, 1976–1987. [Google Scholar] [CrossRef] [PubMed]
- Harper, T.M.; June, C.M.; Taracila, M.A.; Bonomo, R.A.; Powers, R.A.; Leonard, D.A. Multiple substitutions lead to increased loop flexibility and expanded specificity in Acinetobacter baumannii carbapenemase OXA-239. Biochem. J. 2018, 475, 273–288. [Google Scholar] [CrossRef]
- Stewart, N.K.; Smith, C.A.; Antunes, N.T.; Toth, M.; Vakulenko, S.B. Role of the hydrophobic bridge in the carbapenemase activity of class D β-lactamases. Antimicrob. Agents Chemother. 2019, 63, e02191-18. [Google Scholar] [CrossRef]
- Stewart, N.K.; Toth, M.; Alqurafi, M.A.; Chai, W.; Nguyen, T.Q.; Quan, P.; Lee, M.; Buynak, J.D.; Smith, C.A.; Vakulenko, S.B. C6 hydroxymethyl-substituted carbapenem MA-1-206 inhibits the major Acinetobacter baumannii carbapenemase oxa-23 by impeding deacylation. Mbio 2022, 13, e00367-22. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Golemi, D.; Maveyraud, L.; Vakulenko, S.; Samama, J.P.; Mobashery, S. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc. Natl. Acad. Sci. USA 2001, 98, 14280–14285. [Google Scholar] [CrossRef] [PubMed]
- Ringnér, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef]
- Palese, L.L. A random version of principal component analysis in data clustering. Comput. Biol. Chem. 2018, 73, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Santillana, E.; Beceiro, A.; Bou, G.; Romero, A. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc. Natl. Acad. Sci. USA 2007, 104, 5354–5359. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.D.; Calderone, V.; De Luca, F.; Benvenuti, M.; Giuliani, F.; Bellucci, L.; Tafi, A.; Nordmann, P.; Botta, M.; Rossolini, G.M.; et al. Crystal structure of the OXA-48 β-lactamase reveals mechanistic diversity among class D carbapenemases. Chem. Biol. 2009, 16, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Antunes, N.T.; Toth, M.; Vakulenko, S.B. Crystal structure of carbapenemase OXA-58 from Acinetobacter baumannii. Antimicrob. Agents Chemother. 2014, 58, 2135–2143. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Bossis, F.; Palese, L.L. Amyloid beta (1–42) in aqueous environments: Effects of ionic strength and E22Q (Dutch) mutation. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 2486–2493. [Google Scholar] [CrossRef]
- Bossis, F.; De Grassi, A.; Palese, L.L.; Pierri, C.L. Prediction of high-and low-affinity quinol-analogue-binding sites in the aa 3 and bo 3 terminal oxidases from Bacillus subtilis and Escherichia coli. Biochem. J. 2014, 461, 305–314. [Google Scholar] [CrossRef]
- Sardanelli, A.M.; Isgrò, C.; Palese, L.L. SARS-CoV-2 main protease active site ligands in the human metabolome. Molecules 2021, 26, 1409. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Bossis, F.; Palese, L.L. Molecular dynamics in cytochrome c oxidase Mössbauer spectra deconvolution. Biochem. Biophys. Res. Commun. 2011, 404, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Palese, L.L. Correlation analysis of Trp-cage dynamics in folded and unfolded states. J. Phys. Chem. B 2015, 119, 15568–15573. [Google Scholar] [CrossRef]
- Palese, L.L. Random matrix theory in molecular dynamics analysis. Biophys. Chem. 2015, 196, 1–9. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. In Proceedings of the 15th Python IN Science Conferences (SCIPY 2016), Austin, TX, USA, 11–17 July 2016. [Google Scholar]
- Flower, D. Rotational superposition: A review of methods. J. Mol. Graph. Model. 1999, 17, 238–244. [Google Scholar] [PubMed]
- Theobald, D.L. Rapid calculation of RMSDs using a quaternion-based characteristic polynomial. Acta Crystallogr. A 2005, 61, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Agrafiotis, D.K.; Theobald, D.L. Fast determination of the optimal rotational matrix for macromolecular superpositions. J. Comput. Chem. 2010, 31, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Welford, B. Note on a method for calculating corrected sums of squares and products. Technometrics 1962, 4, 419–420. [Google Scholar] [CrossRef]
- Raschka, S. Python Machine Learning; Packt Publishing: Birmingham, UK, 2015. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Jupyter. Available online: https://jupyter.org/ (accessed on 21 December 2022).
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrigoni, R.; Ballini, A.; Santacroce, L.; Palese, L.L. The Dynamics of OXA-23 β-Lactamase from Acinetobacter baumannii. Int. J. Mol. Sci. 2023, 24, 17527. https://doi.org/10.3390/ijms242417527
Arrigoni R, Ballini A, Santacroce L, Palese LL. The Dynamics of OXA-23 β-Lactamase from Acinetobacter baumannii. International Journal of Molecular Sciences. 2023; 24(24):17527. https://doi.org/10.3390/ijms242417527
Chicago/Turabian StyleArrigoni, Roberto, Andrea Ballini, Luigi Santacroce, and Luigi Leonardo Palese. 2023. "The Dynamics of OXA-23 β-Lactamase from Acinetobacter baumannii" International Journal of Molecular Sciences 24, no. 24: 17527. https://doi.org/10.3390/ijms242417527
APA StyleArrigoni, R., Ballini, A., Santacroce, L., & Palese, L. L. (2023). The Dynamics of OXA-23 β-Lactamase from Acinetobacter baumannii. International Journal of Molecular Sciences, 24(24), 17527. https://doi.org/10.3390/ijms242417527