
Dysregulated Cell Homeostasis and miRNAs in Human iPSC-Derived Cardiomyocytes from a Propionic Acidemia Patient with Cardiomyopathy
 , , and
, , and 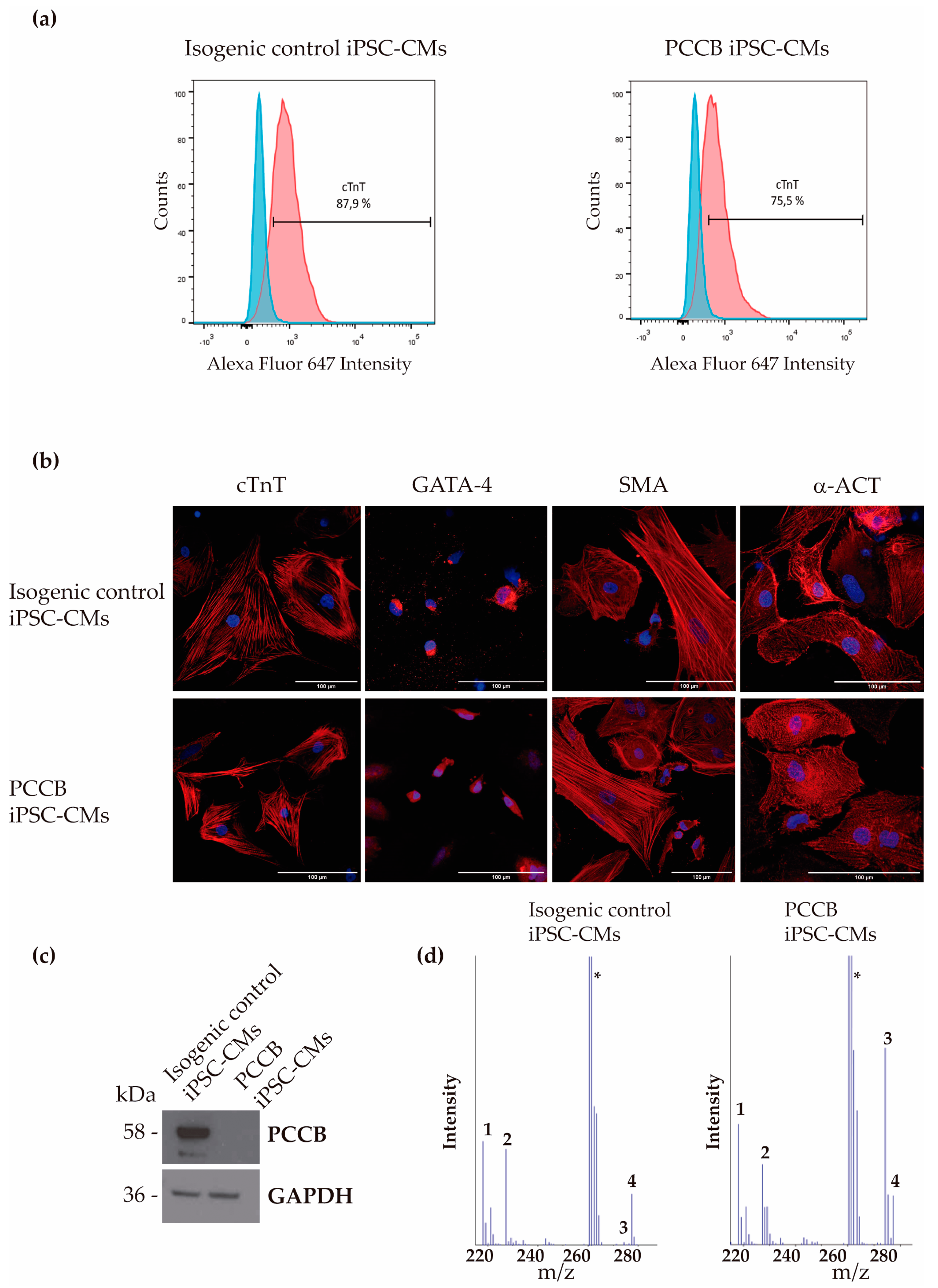{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation and Characterization of iPSC-Derived Cardiomyocytes
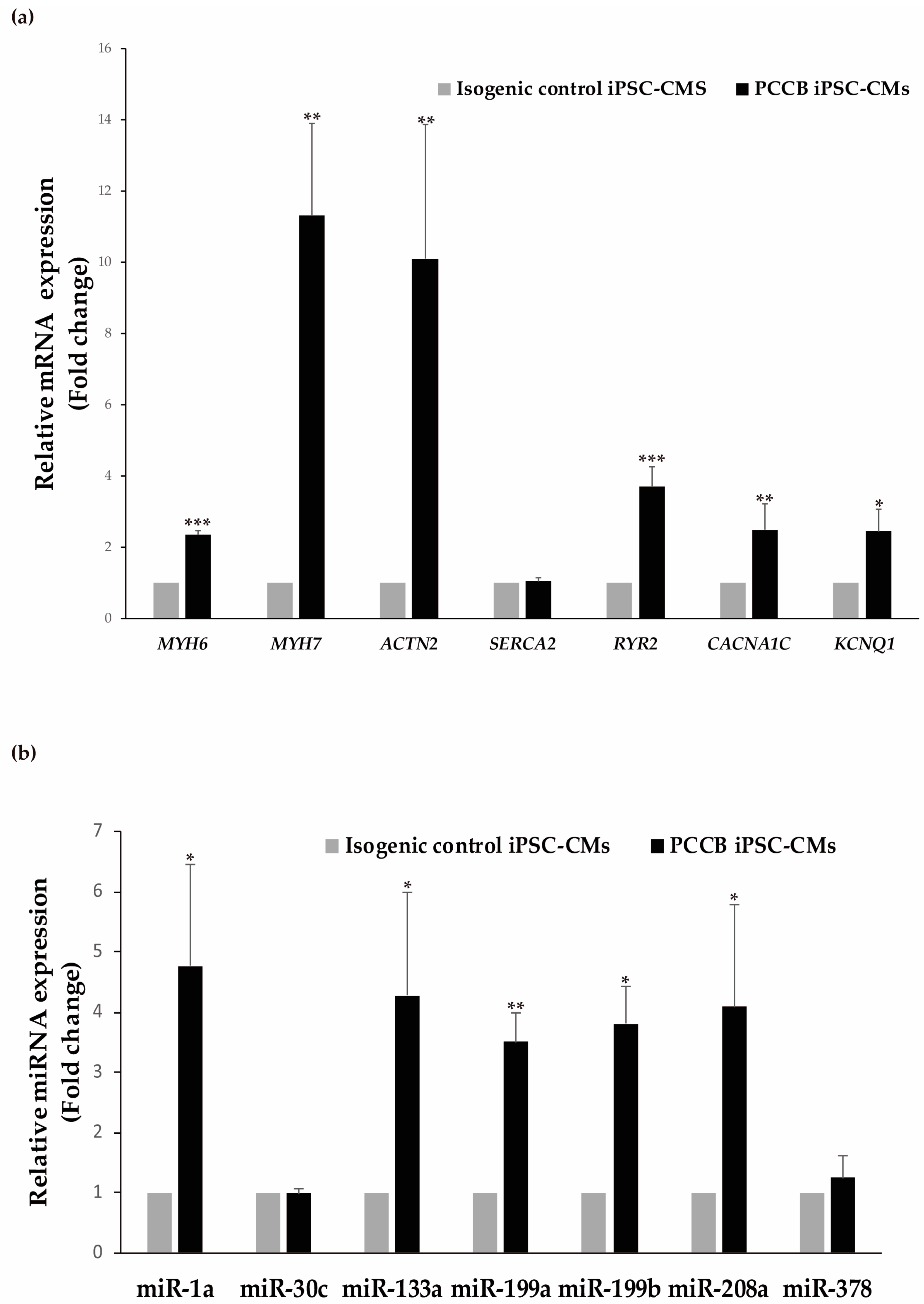
2.2. Evaluation of Cardiac Damage Markers and miRNAs
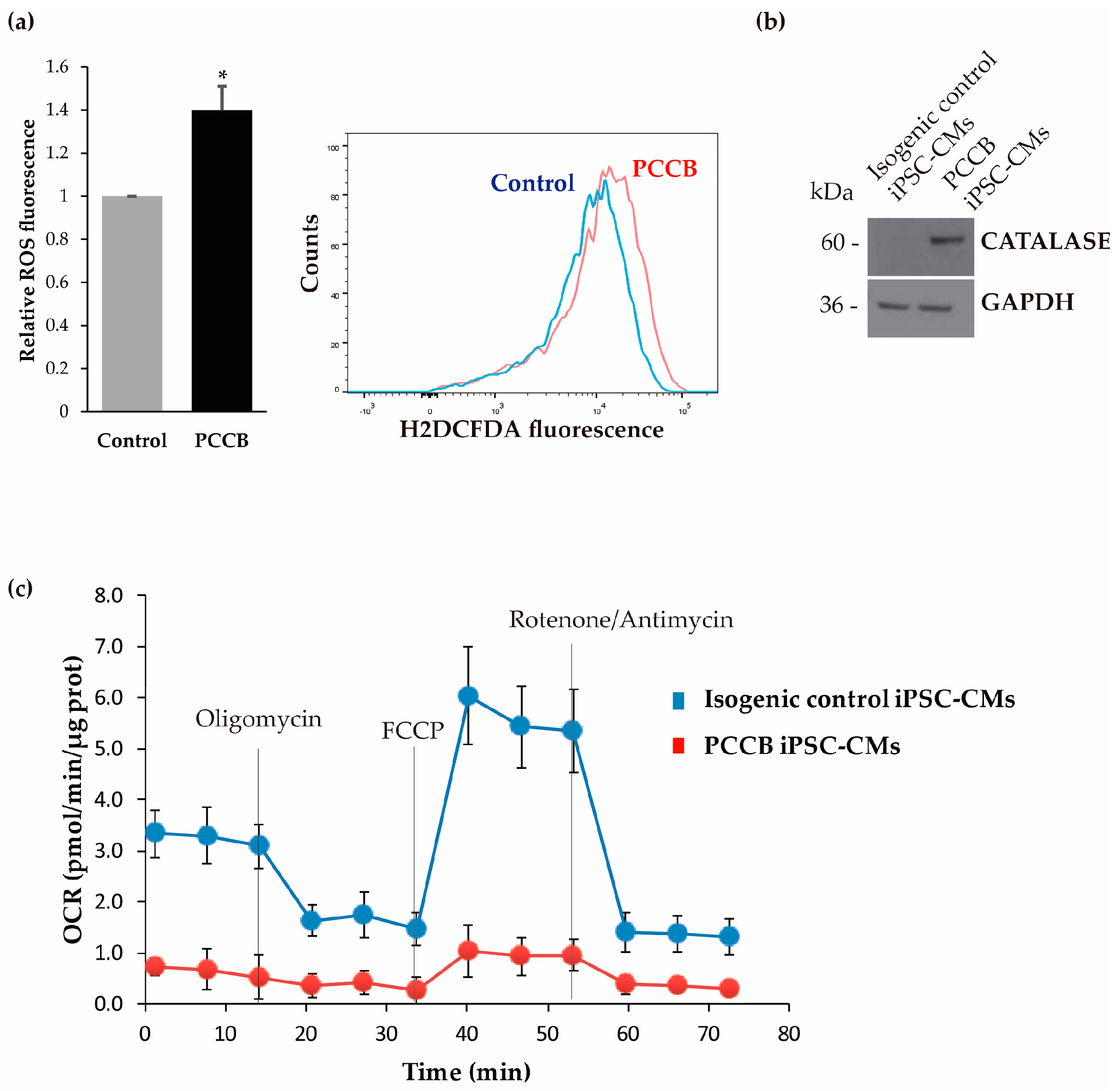
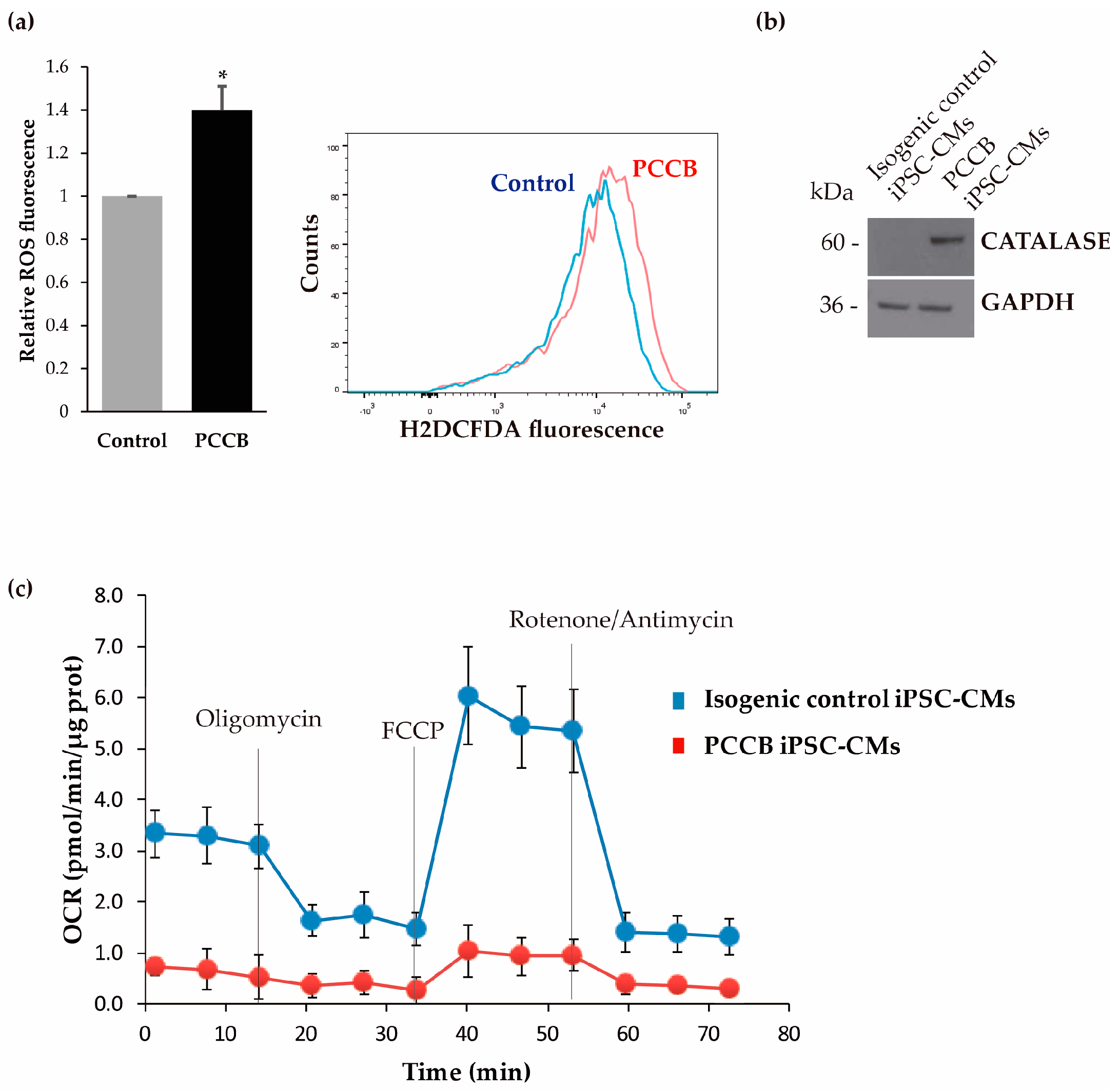
2.3. Analysis of Oxidative Stress and Mitochondrial Function
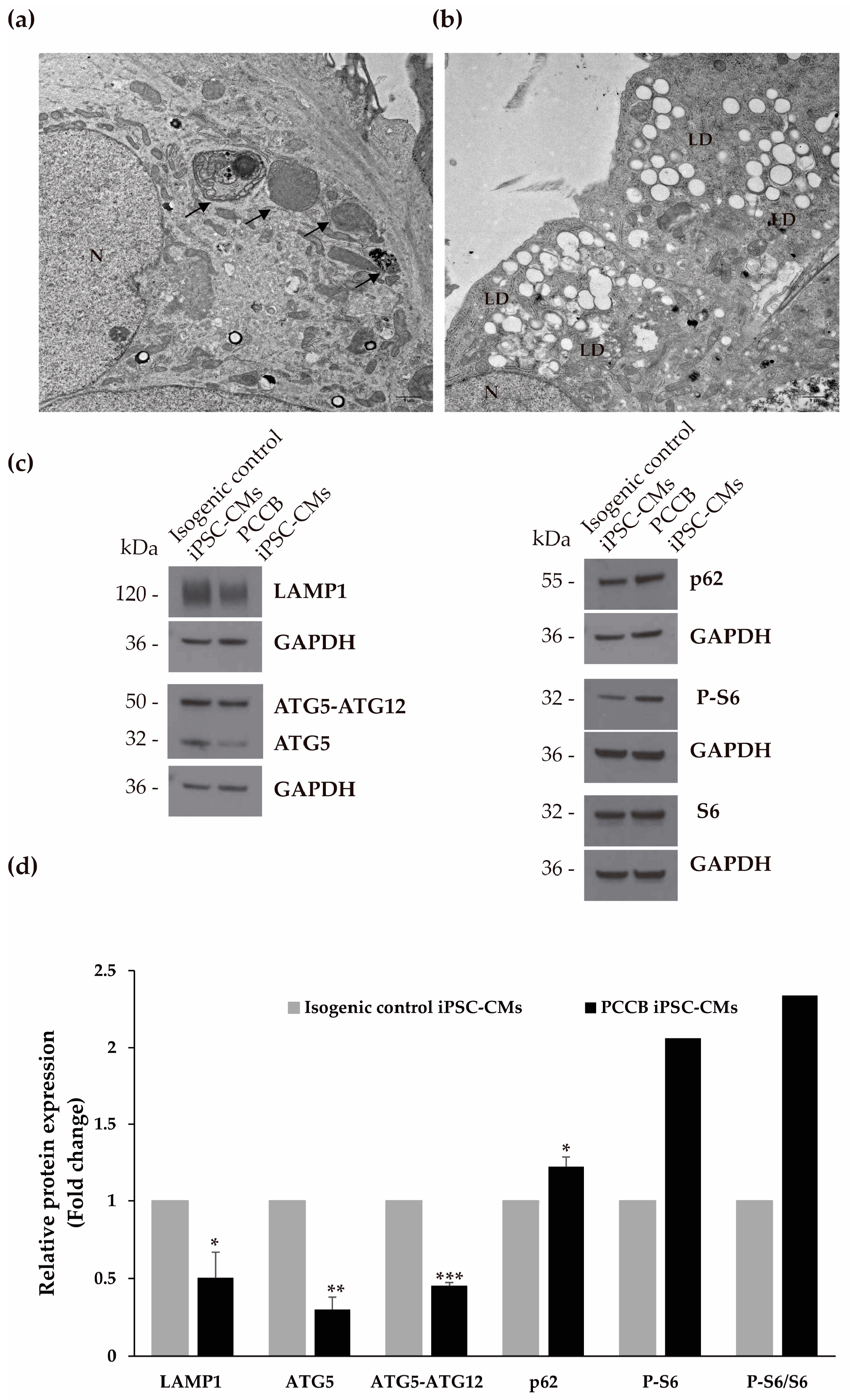
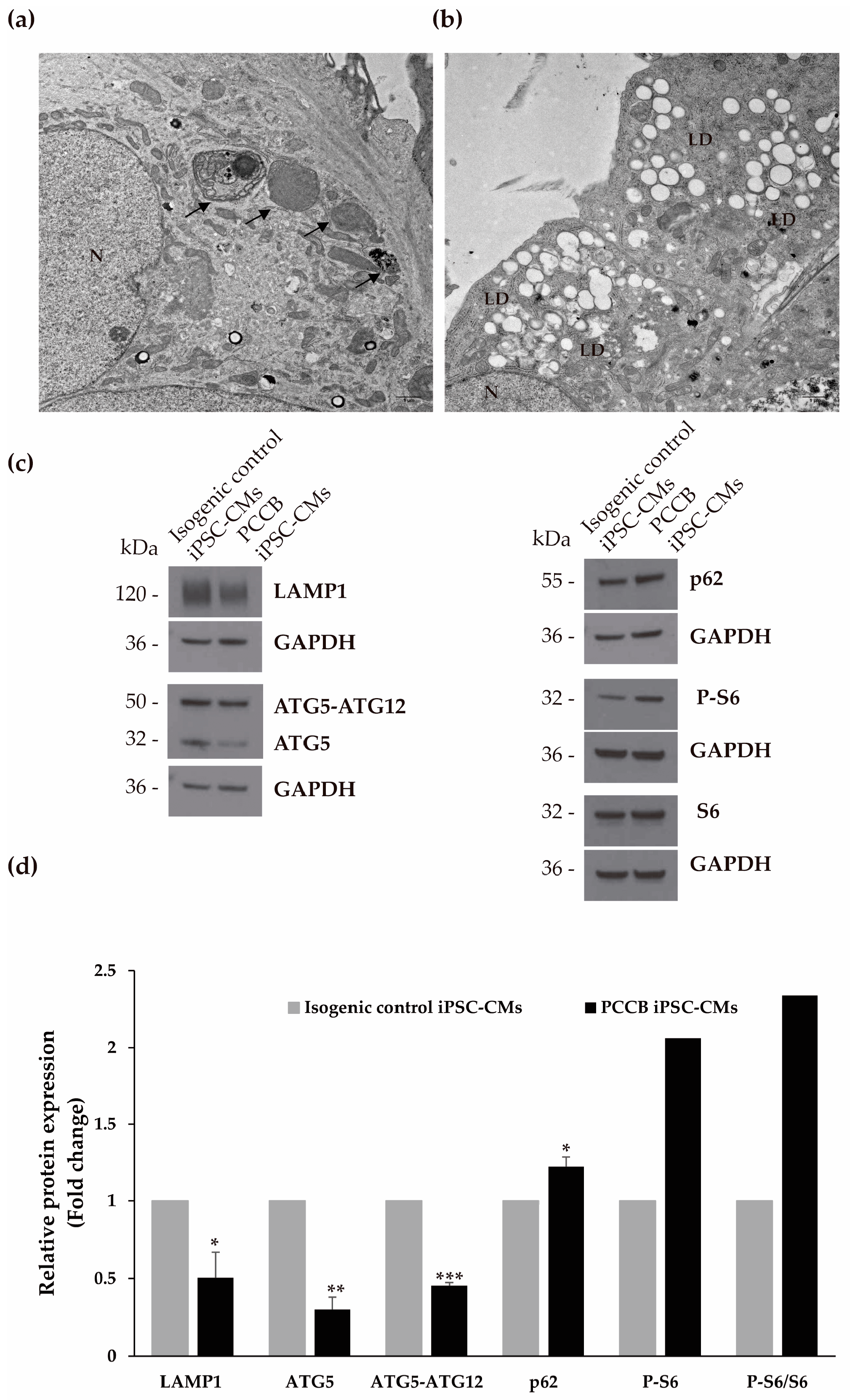
2.4. Ultrastructure Analysis and Evaluation of Autophagy Process
2.5. Evaluation of Protein Expression Involved in Mitochondria-Associated Membranes, Endoplasmic Reticulum Stress, Apoptosis, and Mitochondrial Biogenesis
3. Discussion
4. Materials and Methods
4.1. Maintenance of hiPSC Lines
4.2. Differentiation of hiPSCs into Cardiomyocytes
4.3. Immunofluorescence Staining
4.4. Flow Cytometry
4.5. Quantitative Reverse Transcription PCR (RT-qPCR)
4.6. Western Blotting
4.7. Biochemical Measurements
4.8. Electron Microscopy Sample Preparation and Analysis
4.9. Mitochondrial Bioenergetics Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fenton, W.A.; Gravel, R.A.; Rosenberg, L.E. Disorders of Propionate and Methylmalonate Metabolism. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2165–2190. [Google Scholar]
- Mardach, R.; Verity, M.A.; Cederbaum, S.D. Clinical, Pathological, and Biochemical Studies in a Patient with Propionic Acidemia and Fatal Cardiomyopathy. Mol. Genet. Metab. 2005, 85, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Addonizio, L.J.; Barshop, B.A.; Chung, W.K. Unusual Presentation of Propionic Acidaemia as Isolated Cardiomyopathy. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S97–S101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacevic, A.; Garbade, S.F.; Hoffmann, G.F.; Gorenflo, M.; Kolker, S.; Staufner, C. Cardiac Phenotype in Propionic Acidemia—Results of an Observational Monocentric Study. Mol. Genet. Metab. 2020, 130, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Krywawych, S.; Richard, E.; Desviat, L.R.; Swietach, P. Cardiac Complications of Propionic and Other Inherited Organic Acidemias. Front. Cardiovasc. Med. 2020, 7, 617451. [Google Scholar] [CrossRef]
- Baumgartner, D.; Scholl-Burgi, S.; Sass, J.O.; Sperl, W.; Schweigmann, U.; Stein, J.I.; Karall, D. Prolonged QTc Intervals and Decreased Left Ventricular Contractility in Patients with Propionic Acidemia. J. Pediatr. 2007, 150, 192–197.e1. [Google Scholar] [CrossRef]
- Jameson, E.; Walter, J. Cardiac Arrest Secondary to Long QT(C )in a Child with Propionic Acidemia. Pediatr. Cardiol. 2008, 29, 969–970. [Google Scholar] [CrossRef]
- Guenzel, A.J.; Hofherr, S.E.; Hillestad, M.; Barry, M.; Weaver, E.; Venezia, S.; Kraus, J.P.; Matern, D.; Barry, M.A. Generation of a Hypomorphic Model of Propionic Acidemia Amenable to Gene Therapy Testing. Mol. Ther. 2013, 21, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Fulgencio-Covian, A.; Alonso-Barroso, E.; Guenzel, A.J.; Rivera-Barahona, A.; Ugarte, M.; Perez, B.; Barry, M.A.; Perez-Cerda, C.; Richard, E.; Desviat, L.R. Pathogenic Implications of Dysregulated MiRNAs in Propionic Acidemia Related Cardiomyopathy. Transl. Res. 2020, 218, 43–56. [Google Scholar] [CrossRef]
- Gallego-Villar, L.; Rivera-Barahona, A.; Cuevas-Martin, C.; Guenzel, A.; Perez, B.; Barry, M.A.; Murphy, M.P.; Logan, A.; Gonzalez-Quintana, A.; Martin, M.A.; et al. In Vivo Evidence of Mitochondrial Dysfunction and Altered Redox Homeostasis in a Genetic Mouse Model of Propionic Acidemia: Implications for the Pathophysiology of This Disorder. Free Radic. Biol. Med. 2016, 96, 1–12. [Google Scholar] [CrossRef]
- Tamayo, M.; Fulgencio-Covian, A.; Navarro-Garcia, J.A.; Val-Blasco, A.; Ruiz-Hurtado, G.; Gil-Fernandez, M.; Martin-Nunes, L.; Lopez, J.A.; Desviat, L.R.; Delgado, C.; et al. Intracellular Calcium Mishandling Leads to Cardiac Dysfunction and Ventricular Arrhythmias in a Mouse Model of Propionic Acidemia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165586. [Google Scholar] [CrossRef]
- Alonso-Barroso, E.; Perez, B.; Desviat, L.R.; Richard, E. Cardiomyocytes Derived from Induced Pluripotent Stem Cells as a Disease Model for Propionic Acidemia. Int. J. Mol. Sci. 2021, 22, 1161. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Marquez, A.; Alonso-Barroso, E.; Cerro-Tello, G.; Bravo-Alonso, I.; Arribas-Carreira, L.; Briso-Montiano, A.; Navarrete, R.; Perez-Cerda, C.; Ugarte, M.; Perez, B.; et al. Generation and Characterization of a Human IPSC Line (UAMi004-A) from a Patient with Propionic Acidemia Due to Defects in the PCCB Gene. Stem Cell Res. 2019, 38, 101469. [Google Scholar] [CrossRef] [PubMed]
- Fulgencio-Covian, A.; Alvarez, M.; Pepers, B.A.; Lopez-Marquez, A.; Ugarte, M.; Perez, B.; van Roon-Mom, W.M.C.; Desviat, L.R.; Richard, E. Generation of a Gene-Corrected Human Isogenic Line (UAMi006-A) from Propionic Acidemia Patient IPSC with an Homozygous Mutation in the PCCB Gene Using CRISPR/Cas9 Technology. Stem Cell Res. 2020, 49, 102055. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-Specific MiRNA in Cardiogenesis, Heart Function, and Cardiac Pathology (with Focus on Myocardial Infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.-H.; Tatsuguchi, M.; Huang, Z.-P.; Chen, J.-F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a Is a Regulator of Cardiac Hypertrophy and Conduction in Mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, A.; Balizadeh Karami, A.R.; Dehghan Mashtani, V.; Sahraei, T.; Bandani Tarashoki, Z.; Khattavian, E.; Mobarak, S.; Moradi Kazerouni, H.; Radmanesh, E. Evaluation of Oxidative Stress, Apoptosis, and Expression of MicroRNA-208a and MicroRNA-1 in Cardiovascular Patients. Rep. Biochem. Mol. Biol. 2021, 10, 183–196. [Google Scholar] [CrossRef]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. MiR-199a Impairs Autophagy and Induces Cardiac Hypertrophy through MTOR Activation. Cell Death Differ. 2017, 24, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- el Azzouzi, H.; Leptidis, S.; Dirkx, E.; Hoeks, J.; van Bree, B.; Brand, K.; McClellan, E.A.; Poels, E.; Sluimer, J.C.; van den Hoogenhof, M.M.; et al. The Hypoxia-Inducible MicroRNA Cluster MiR-199a-214 Targets Myocardial PPARdelta and Impairs Mitochondrial Fatty Acid Oxidation. Cell Metab. 2013, 18, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Juárez-Rojas, J.G.; Reyes-Soffer, G.; Conlon, D.; Ginsberg, H.N. Autophagy and Cardiometabolic Risk Factors. Rev. Endocr. Metab. Disord. 2014, 15, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Li, M.-Y.; Li, P.-F.; Cao, J.-M. MicroRNAs in Cardiac Autophagy: Small Molecules and Big Role. Cells 2018, 7, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, H.-Y.; Shao, J.; Zhu, L.; Xie, T.-H.; Cai, J.; Wang, W.; Cai, M.-X.; Wang, Z.-L.; Yao, Y.; et al. GRP75 Modulates Endoplasmic Reticulum–Mitochondria Coupling and Accelerates Ca2+-Dependent Endothelial Cell Apoptosis in Diabetic Retinopathy. Biomolecules 2022, 12, 1778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jia, W.-K.; Jian, Z.; Zhao, L.; Liu, C.-C.; Wang, Y.; Xiao, Y.-B. Downregulation of MicroRNA-199a-5p Protects Cardiomyocytes in Cyanotic Congenital Heart Disease by Attenuating Endoplasmic Reticulum Stress. Mol. Med. Rep. 2017, 16, 2992–3000. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.-I.; Sabry, A.D.; Cawley, K.M.; Warren, J.S. Multiple Levels of PGC-1α Dysregulation in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piquereau, J.; Moulin, M.; Zurlo, G.; Mateo, P.; Gressette, M.; Paul, J.-L.; Lemaire, C.; Ventura-Clapier, R.; Veksler, V.; Garnier, A. Cobalamin and Folate Protect Mitochondrial and Contractile Functions in a Murine Model of Cardiac Pressure Overload. J. Mol. Cell. Cardiol. 2017, 102, 34–44. [Google Scholar] [CrossRef]
- Pereira, R.O.; Wende, A.R.; Crum, A.; Hunter, D.; Olsen, C.D.; Rawlings, T.; Riehle, C.; Ward, W.F.; Abel, E.D. Maintaining PGC-1α Expression Following Pressure Overload-Induced Cardiac Hypertrophy Preserves Angiogenesis but Not Contractile or Mitochondrial Function. FASEB J. 2014, 28, 3691–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, M.; Giordano, C.; Nediani, C.; Travaglini, C.; Borchi, E.; Zani, M.; Feccia, M.; Mancini, M.; Petrozza, V.; Cossarizza, A.; et al. Induction of Mitochondrial Biogenesis Is a Maladaptive Mechanism in Mitochondrial Cardiomyopathies. J. Am. Coll. Cardiol. 2007, 50, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA Replication Impairs Mitochondrial Biogenesis in Human Failing Hearts. Circ. Res. 2010, 106, 1541–1548. [Google Scholar] [CrossRef] [Green Version]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1α and ERRα Target Gene Downregulation Is a Signature of the Failing Human Heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Duygu, B.; Poels, E.M.; Juni, R.; Bitsch, N.; Ottaviani, L.; Olieslagers, S.; de Windt, L.J.; da Costa Martins, P.A. MiR-199b-5p Is a Regulator of Left Ventricular Remodeling Following Myocardial Infarction. Non-Coding RNA Res. 2017, 2, 18–26. [Google Scholar] [CrossRef]
- Belevych, A.E.; Sansom, S.E.; Terentyeva, R.; Ho, H.-T.; Nishijima, Y.; Martin, M.M.; Jindal, H.K.; Rochira, J.A.; Kunitomo, Y.; Abdellatif, M.; et al. MicroRNA-1 and -133 Increase Arrhythmogenesis in Heart Failure by Dissociating Phosphatase Activity from RyR2 Complex. PLoS ONE 2011, 6, e28324. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Belevych, A.E.; Terentyeva, R.; Martin, M.M.; Malana, G.E.; Kuhn, D.E.; Abdellatif, M.; Feldman, D.S.; Elton, T.S.; Györke, S. MiR-1 Overexpression Enhances Ca(2+) Release and Promotes Cardiac Arrhythmogenesis by Targeting PP2A Regulatory Subunit B56alpha and Causing CaMKII-Dependent Hyperphosphorylation of RyR2. Circ. Res. 2009, 104, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Zhao, J.; Tuazon, J.P.; Borlongan, C.V.; Yu, G. MicroRNA-133a and Myocardial Infarction. Cell Transplant. 2019, 28, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boczek, N.J.; Ye, D.; Jin, F.; Tester, D.J.; Huseby, A.; Bos, J.M.; Johnson, A.J.; Kanter, R.; Ackerman, M.J. Identification and Functional Characterization of a Novel CACNA1C-Mediated Cardiac Disorder Characterized by Prolonged QT Intervals with Hypertrophic Cardiomyopathy, Congenital Heart Defects, and Sudden Cardiac Death. Circ. Arrhythm. Electrophysiol. 2015, 8, 1122–1132. [Google Scholar] [CrossRef] [Green Version]
- Good, J.-M.; Fellmann, F.; Bhuiyan, Z.A.; Rotman, S.; Pruvot, E.; Schläpfer, J. ACTN2 Variant Associated with a Cardiac Phenotype Suggestive of Left-Dominant Arrhythmogenic Cardiomyopathy. Heart Case Rep. 2020, 6, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chen, D.-F.; Dong, H.-L.; Liu, G.-L.; Wu, Z.-Y. A Novel Frameshift ACTN2 Variant Causes a Rare Adult-Onset Distal Myopathy with Multi-Minicores. CNS Neurosci. Ther. 2021, 27, 1198–1205. [Google Scholar] [CrossRef]
- Bodi, I.; Grunert, S.C.; Becker, N.; Stoelzle-Feix, S.; Spiekerkoetter, U.; Zehender, M.; Bugger, H.; Bode, C.; Odening, K.E. Mechanisms of Acquired Long QT Syndrome in Patients with Propionic Academia. Heart Rhythm 2016, 13, 1335–1345. [Google Scholar] [CrossRef]
- Wang, Y.; Zankov, D.P.; Jiang, M.; Zhang, M.; Henderson, S.C.; Tseng, G.-N. [Ca2+]i Elevation and Oxidative Stress Induce KCNQ1 Protein Translocation from the Cytosol to the Cell Surface and Increase Slow Delayed Rectifier (IKs) in Cardiac Myocytes. J. Biol. Chem. 2013, 288, 35358–35371. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, Z.; Dong, H.; Ding, Y.; He, R.; Kang, L.; Li, D.; Shen, M.; Jin, Y.; Zhang, Y.; et al. Analysis of the Relationship between Phenotypes and Genotypes in 60 Chinese Patients with Propionic Acidemia: A Fourteen-Year Experience at a Tertiary Hospital. Orphanet J. Rare Dis. 2022, 17, 135. [Google Scholar] [CrossRef]
- Wang, J.; Liew, O.W.; Richards, A.M.; Chen, Y.T. Overview of MicroRNAs in Cardiac Hypertrophy, Fibrosis, and Apoptosis. Int. J. Mol. Sci. 2016, 17, 749. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for MicroRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, E.; Jorge-Finnigan, A.; Garcia-Villoria, J.; Merinero, B.; Desviat, L.R.; Gort, L.; Briones, P.; Leal, F.; Perez-Cerda, C.; Ribes, A.; et al. Genetic and Cellular Studies of Oxidative Stress in Methylmalonic Aciduria (MMA) Cobalamin Deficiency Type C (CblC) with Homocystinuria (MMACHC). Hum. Mutat. 2009, 30, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, S.; Belanger-Quintana, A.; Fernández-Felix, B.M.; Pérez-Cerdá, C.; Merinero, B.; Ruiz-Sala, P.; Arrieta, F.; Martínez-Pardo, M. Long-term Follow-up with Filter Paper Samples in Patients with Propionic Acidemia. JIMD Rep. 2020, 57, 44–51. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez, M.; Ruiz-Sala, P.; Pérez, B.; Desviat, L.R.; Richard, E. Dysregulated Cell Homeostasis and miRNAs in Human iPSC-Derived Cardiomyocytes from a Propionic Acidemia Patient with Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 2182. https://doi.org/10.3390/ijms24032182
Álvarez M, Ruiz-Sala P, Pérez B, Desviat LR, Richard E. Dysregulated Cell Homeostasis and miRNAs in Human iPSC-Derived Cardiomyocytes from a Propionic Acidemia Patient with Cardiomyopathy. International Journal of Molecular Sciences. 2023; 24(3):2182. https://doi.org/10.3390/ijms24032182
Chicago/Turabian StyleÁlvarez, Mar, Pedro Ruiz-Sala, Belén Pérez, Lourdes Ruiz Desviat, and Eva Richard. 2023. "Dysregulated Cell Homeostasis and miRNAs in Human iPSC-Derived Cardiomyocytes from a Propionic Acidemia Patient with Cardiomyopathy" International Journal of Molecular Sciences 24, no. 3: 2182. https://doi.org/10.3390/ijms24032182