
PLEKHM2 Loss of Function Impairs the Activity of iPSC-Derived Neurons via Regulation of Autophagic Flux
 , , and
, , and
Abstract
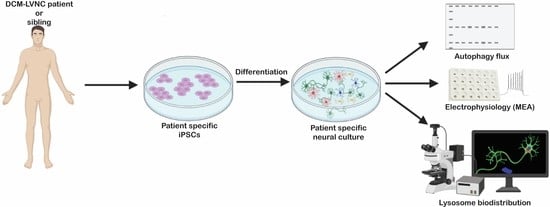
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. PLEKHM2[delAG] Patients and iPSCs
2.2. PLEKHM2[delAG] iPSCs Can Differentiate into iMNs
2.3. PLEKHM2[delAG] Cultures Show Reduced Autophagic Activity
2.4. PLEKHM2[delAG] Culture Activity Peaks Later Than Its Control
2.5. The Mutation in PLEKHM2 Leads to Lysosomal Dysfunction
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Neuronal Differentiation
4.3. Flow Cytometry Quantification
4.4. Autophagy Flux Detection
4.5. Microelectrode Array (MEA) Assay
4.6. Western Blot
4.7. Immunocytochemistry
4.8. Biodistribution Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of Lysosomes in Cellular Autophagy Induced in Rat Liver by Glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy: From Phenomenology to Molecular Understanding in Less Than. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Puri, C.; Moreau, K.; Rubinsztein, D.C. The Role of Membrane-Trafficking Small GTPases in the Regulation of Autophagy. J. Cell Sci. 2013, 126, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and Diversity in Autophagy Mechanisms: Lessons from Yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, E.; Levitas, A.; Singh, S.R.; Braiman, A.; Ofir, R.; Etzion, S.; Sheffield, V.C.; Etzion, Y.; Carrier, L.; Parvari, R. PLEKHM2 Mutation Leads to Abnormal Localization of Lysosomes, Impaired Autophagy Flux and Associates with Recessive Dilated Cardiomyopathy and Left Ventricular Noncompaction. Hum. Mol. Genet. 2015, 24, 7227–7240. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Ferreira, C.; Munro, S. Arl8 and SKIP Act Together to Link Lysosomes to Kinesin-1. Dev. Cell 2011, 21, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and Functions of Lysosome Positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [Green Version]
- Keren-Kaplan, T.; Bonifacino, J.S. ARL8 Relieves SKIP Autoinhibition to Enable Coupling of Lysosomes to Kinesin-1. Curr. Biol. 2021, 31, 540–554.e5. [Google Scholar] [CrossRef]
- De Pace, R.; Britt, D.J.; Mercurio, J.; Foster, A.M.; Djavaherian, L.; Hoffmann, V.; Abebe, D.; Bonifacino, J.S. Synaptic Vesicle Precursors and Lysosomes Are Transported by Different Mechanisms in the Axon of Mammalian Neurons. Cell Rep. 2020, 31, 107775. [Google Scholar] [CrossRef]
- Samie, M.; Cresswell, P. The Transcription Factor TFEB Acts as a Molecular Switch That Regulates Exogenous Antigen-Presentation Pathways. Nat. Immunol. 2015, 16, 729–736. [Google Scholar] [CrossRef]
- Kruse, K.B.; Dear, A.; Kaltenbrun, E.R.; Crum, B.E.; George, P.M.; Brennan, S.O.; McCracken, A.A. Mutant Fibrinogen Cleared from the Endoplasmic Reticulum via Endoplasmic Reticulum-Associated Protein Degradation and Autophagy: An Explanation for Liver Disease. Am. J. Pathol. 2006, 168, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, M.; Carnio, S.; Labeit, D.; Sandri, M. Role of Autophagy, SQSTM1, SH3GLB1, and TRIM63 in the Turnover of Nicotinic Acetylcholine Receptors. Autophagy 2014, 10, 123–136. [Google Scholar] [CrossRef]
- Shen, D.N.; Zhang, L.H.; Wei, E.Q.; Yang, Y. Autophagy in Synaptic Development, Function, and Pathology. Neurosci. Bull. 2015, 31, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Varo, R.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Baglietto-Vargas, D.; Moreno-Gonzalez, I.; De Castro, V.; Jimenez, S.; Ruano, D.; Vizuete, M.; et al. Abnormal Accumulation of Autophagic Vesicles Correlates with Axonal and Synaptic Pathology in Young Alzheimer’s Mice Hippocampus. Acta Neuropathol. 2012, 123, 53–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seranova, E.; Palhegyi, A.M.; Verma, S.; Dimova, S.; Lasry, R.; Naama, M.; Sun, C.; Barrett, T.; Rosenstock, T.R.; Kumar, D.; et al. Human Induced Pluripotent Stem Cell Models of Neurodegenerative Disorders for Studying the Biomedical Implications of Autophagy. J. Mol. Biol. 2020, 432, 2754–2798. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-Mediated Neuroprotection in the MPTP Mouse Model of Parkinson’s Disease: Critical Role for the Astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Danés, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jiménez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-Specific Phenotypes in Dopamine Neurons from Human IPS-Based Models of Genetic and Sporadic Parkinson’s Disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef]
- Corrionero, A.; Horvitz, H.R. A C9orf72 ALS/FTD Ortholog Acts in Endolysosomal Degradation and Lysosomal Homeostasis. Curr. Biol. 2018, 28, 1522–1535.e5. [Google Scholar] [CrossRef]
- Farías, G.G.; Guardia, C.M.; De Pace, R.; Britt, D.J.; Bonifacino, J.S. BORC/Kinesin-1 Ensemble Drives Polarized Transport of Lysosomes into the Axon. Proc. Natl. Acad. Sci. USA 2017, 114, E2955–E2964. [Google Scholar] [CrossRef] [Green Version]
- Roney, J.C.; Li, S.; Farfel-Becker, T.; Huang, N.; Sun, T.; Xie, Y.; Cheng, X.T.; Lin, M.Y.; Platt, F.M.; Sheng, Z.H. Lipid-Mediated Motor-Adaptor Sequestration Impairs Axonal Lysosome Delivery Leading to Autophagic Stress and Dystrophy in Niemann-Pick Type C. Dev. Cell 2021, 56, 1452–1468.e8. [Google Scholar] [CrossRef] [PubMed]
- Ben-zvi, H.; Korover, N.; Rabinski, T.; Ofir, R.; Cohen, S. Generation and Characterization of Three Human Induced Pluripotent Stem Cell Lines ( IPSC ) from Two Family Members with Dilated Cardiomyopathy and Left Ventricular Noncompaction ( DCM-LVNC ) and One Healthy Heterozygote Sibling. Stem Cell Res. 2021, 53, 102382. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of Autophagy in the Central Nervous System Causes Neurodegeneration in Mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 Activation in Astrocytes Protects against Neurodegeneration in Mouse Models of Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Barthakur, S.; Kasendra, M.; Lucchesi, C.; Kerns, J.; et al. Human IPSC-Derived Blood-Brain Barrier Chips Enable Disease Modeling and Personalized Medicine Applications. Cell Stem Cell 2019, 24, 995–1005.e6. [Google Scholar] [CrossRef]
- Laperle, A.H.; Sances, S.; Yucer, N.; Dardov, V.J.; Garcia, V.J.; Ho, R.; Fulton, A.N.; Jones, M.R.; Roxas, K.M.; Avalos, P.; et al. IPSC Modeling of Young-Onset Parkinson’s Disease Reveals a Molecular Signature of Disease and Novel Therapeutic Candidates. Nat. Med. 2020, 26, 289–299. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of Basal Autophagy in Neural Cells Causes Neurodegenerative Disease in Mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Young, J.E.; Martinez, R.A.; La Spada, A.R. Nutrient Deprivation Induces Neuronal Autophagy and Implicates Reduced Insulin Signaling in Neuroprotective Autophagy Activation. J. Biol. Chem. 2009, 284, 2363–2373. [Google Scholar] [CrossRef] [Green Version]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes Initiate Distally and Mature during Transport toward the Cell Soma in Primary Neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Black, B.J.; Atmaramani, R.; Pancrazio, J.J. Spontaneous and Evoked Activity from Murine Ventral Horn Cultures on Microelectrode Arrays. Front. Cell. Neurosci. 2017, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewett, K.A.; Christian, C.A.; Bacos, J.T.; Lee, K.Y.; Zhu, J.; Tsai, N.P. Feedback Modulation of Neural Network Synchrony and Seizure Susceptibility by Mdm2-P53-Nedd4-2 Signaling. Mol. Brain 2016, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.W.; Murphy, T.L.; Wlllingham, M.C.; Pastan, I.; August, J.T. Identification of Two Lysosomal Membrane Glycoproteins. J. Cell Biol. 1985, 101, 85–95. [Google Scholar] [CrossRef]
- Unsain, N.; Kononenko, N.L.; Telpoukhovskaia, M.; Givogri, M.I.; Gowrishankar, S.; Cologna, S.M.; Alford, S.T.; Bongarzone, E.R.; Rebiai, R.; Cologna, S.M. Synaptic Function and Dysfunction in Lysosomal Storage Diseases. Front. Cell. Neurosci. 2021, 15, 55. [Google Scholar] [CrossRef]
- De Araujo, M.E.G.; Liebscher, G.; Hess, M.W.; Huber, L.A. Lysosomal Size Matters. Traffic 2020, 21, 60–75. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.; Di Ronza, A.; Lotfi, P.; Sardiello, M. Lysosomes and Brain Health. Annu. Rev. Neurosci. 2018, 41, 255–276. [Google Scholar] [CrossRef]
- Oyarzún, J.E.; Lagos, J.; Vázquez, M.C.; Valls, C.; De la Fuente, C.; Yuseff, M.I.; Alvarez, A.R.; Zanlungo, S. Lysosome Motility and Distribution: Relevance in Health and Disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 1076–1087. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia Clear Neuron-Released α-Synuclein via Selective Autophagy and Prevent Neurodegeneration. Nat. Commun. 2020, 11, 1–14T. [Google Scholar] [CrossRef]
- Lin, M.W.; Lin, C.C.; Chen, Y.H.; Yang, H.B.; Hung, S.Y. Celastrol Inhibits Dopaminergic Neuronal Death of Parkinson’S Disease through Activating Mitophagy. Antioxidants 2020, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s IPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Song, H.; Byun, J.; Park, K.S.; Jang, H.C.; Park, Y.J.; Mook-Jung, I. Accumulation of Autophagosomes Contributes to Enhanced Amyloidogenic APP Processing under Insulin-Resistant Conditions. Autophagy 2012, 8, 1842–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Yin, X.L.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and Amyloid Beta-Induced Defective Autophagy, Mitophagy, Mitochondrial Structural and Functional Changes and Synaptic Damage in Hippocampal Neurons from Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Klein, J.D.; Du, J.; Franch, H.A.; Zhang, L.; Hassounah, F.; Hudson, M.B.; Wang, X.H. Chronic Kidney Disease Induces Autophagy Leading to Dysfunction of Mitochondria in Skeletal Muscle. Am. J. Physiol.-Ren. Physiol. 2017, 312, F1128–F1140. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial Cell Mitochondrial Dysfunction and PINK1 Are Induced by Transforming Growth Factor- Beta1 in Pulmonary Fibrosis. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Han, K.; Kim, S.H.; Choi, M. Computational Modeling of the Effects of Autophagy on Amyloid-β Peptide Levels. Theor. Biol. Med. Model. 2020, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, B.; Liu, L.; Zhang, L.; Ma, T.; Guo, L. Nicotinamide Adenine Dinucleotide Treatment Alleviates the Symptoms of Experimental Autoimmune Encephalomyelitis by Activating Autophagy and Inhibiting the NLRP3 Inflammasome. Int. Immunopharmacol. 2021, 90, 107092. [Google Scholar] [CrossRef]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.M.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired Autophagy in the Lipid-Storage Disorder Niemann-Pick Type C1 Disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Maetzel, D.; Korolchuk, V.I.; Jaenisch, R. Restarting Stalled Autophagy a Potential Therapeutic Approach for the Lipid Storage Disorder, Niemann-Pick Type C1 Disease. Autophagy 2014, 10, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, Y.; Bueno, M.; Ramirez, R.; Álvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. MTORC1 Activation Decreases Autophagy in Aging and Idiopathic Pulmonary Fibrosis and Contributes to Apoptosis Resistance in IPF Fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-Dependent Neurodegeneration and Organelle Transport Deficiencies in Mutant TDP43 Patient-Derived Neurons Are Independent of TDP43 Aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a Promoter of Longevity: Insights from Model Organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Powers, R.W.; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Cell Biology: Regulation of Yeast Replicative Life Span by TOR and Sch9 Response to Nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [Green Version]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in Mice Activates Autophagy and Extends Lifespan. Nat. Commun. 2013, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and Lifespan Extension by the Natural Polyamine Spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Dave Sharp, Z.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Erby Wilkinson, J.; Frenkel, K.; Carter, C.S.; et al. Rapamycin Fed Late in Life Extends Lifespan in Genetically Heterogeneous Mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Canfield, S.G.; Stebbins, M.J.; Morales, B.S.; Asai, S.W.; Vatine, G.D.; Svendsen, C.N.; Palecek, S.P.; Shusta, E.V. An Isogenic Blood–Brain Barrier Model Comprising Brain Endothelial Cells, Astrocytes, and Neurons Derived from Human Induced Pluripotent Stem Cells. J. Neurochem. 2017, 140, 874–888. [Google Scholar] [CrossRef]
- Schevon, C.A.; Weiss, S.A.; McKhann, G.; Goodman, R.R.; Yuste, R.; Emerson, R.G.; Trevelyan, A.J. Evidence of an Inhibitory Restraint of Seizure Activity in Humans. Nat. Commun. 2012, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnatkovsky, V.; Librizzi, L.; Trombin, F.; De Curtis, M. Fast Activity at Seizure Onset Is Mediated by Inhibitory Circuits in the Entorhinal Cortex in Vitro. Ann. Neurol. 2008, 64, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Xiao, G.; Song, Y.; Zhang, Y.; Xing, Y.; Xu, S.; Lu, Z.; Wang, M. Cellular-Scale Microelectrode Arrays to Monitor Movement-Related Neuron Activities in the Epileptic Hippocampus of Awake Mice. IEEE Trans. Biomed. Eng. 2020, 68, 19–25. [Google Scholar] [CrossRef]
- Alsaqati, M.; Heine, V.M.; Harwood, A.J. Pharmacological Intervention to Restore Connectivity Deficits of Neuronal Networks Derived from ASD Patient IPSC with a TSC2 Mutation. Mol. Autism 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S.; Kullmann, D.M.; Kaeser, P.S. Chapter 15—Release of Neurotransmitters. In From Molecules to Networks; Byrne, J.H., Heidelberger, R., Waxham, M.N., Eds.; Academic Press: Boston, USA, 2014; pp. 443–488. [Google Scholar] [CrossRef]
- Pereda, A.E.; Purpura, D.P. Electrical Synapses and Their Functional Interactions with Chemical Synapses. Nat. Rev. Neurosci. 2014, 15, 250–263. [Google Scholar] [CrossRef]
- Cotman, C.W.; McGaugh, J.L. 5—Synaptic Transmission; Cotman, C.W., McGaugh, J.L.., Eds.; Academic Press: New York, NY, USA, 1980; pp. 151–208. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Y.; Li, M.; Zochowski, M.; Budak, M. Synaptic Failure Differentially Affects Pattern Formation in Heterogenous Networks. Front. Neural Circuits 2019, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Taoufik, E.; Kouroupi, G.; Zygogianni, O.; Matsas, R. Synaptic Dysfunction in Neurodegenerative and Neurodevelopmental Diseases: An Overview of Induced Pluripotent Stem-Cell-Based Disease Models. Open Biol. 2018, 8, 180138. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of Autophagy and Reformation of Lysosomes Regulated by MTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic Lysosome Reformation Dysfunction in Glucocerebrosidase Deficient Cells: Relevance to Parkinson Disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef] [Green Version]
- Ohkuma, S.; Poole, B. Cytoplasmic Vacuolation of Mouse Peritoneal Macrophages and the Uptake into Lysosomes of Weakly Basic Substances. J. Cell Biol. 1981, 90, 656–664. [Google Scholar] [CrossRef]
- Johnson, D.E.; Ostrowski, P.; Jaumouillé, V.; Grinstein, S. The Position of Lysosomes within the Cell Determines Their Luminal PH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M. The Role of Autophagy in Cardiomyocytes in the Basal State and in Response to Hemodynamic Stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential Role for Autophagy Protein Atg7 in the Maintenance of Axonal Homeostasis and the Prevention of Axonal Degeneration. Proc. Natl. Acad. Sci. 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal Positioning Coordinates Cellular Nutrient Responses. Nat. Cell Biol. 2011, 13, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.E.; Huo, L.; Maeder, C.I.; Feng, W.; Shen, K. The Balance between Capture and Dissociation of Presynaptic Proteins Controls the Spatial Distribution of Synapses. Neuron 2013, 78, 994–1011. [Google Scholar] [CrossRef] [Green Version]
- Klassen, M.P.; Wu, Y.E.; Maeder, C.I.; Nakae, I.; Cueva, J.G.; Lehrman, E.K.; Tada, M.; Gengyo-Ando, K.; Wang, G.J.; Goodman, M.; et al. An Arf-like Small G Protein, ARL-8, Promotes the Axonal Transport of Presynaptic Cargoes by Suppressing Vesicle Aggregation. Neuron 2010, 66, 710–723. [Google Scholar] [CrossRef] [Green Version]
- Vukoja, A.; Rey, U.; Petzoldt, A.G.; Ott, C.; Vollweiter, D.; Quentin, C.; Puchkov, D.; Reynolds, E.; Lehmann, M.; Hohensee, S.; et al. Presynaptic Biogenesis Requires Axonal Transport of Lysosome-Related Vesicles. Neuron 2018, 99, 1216–1232.e7. [Google Scholar] [CrossRef]
- Missler, M.; Wolff, A.; Merker, H.-J.; Wolff, J.R. Pre- and Postnatal Development of the Primary Visual Cortex of the Common Marmoset. II. Formation, Remodelling, and Elimination of Synapses as Overlapping Processes. J. Comp. Neurol. 1993, 333, 53–67. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W.; Haven, N. By Ca2+ -Regulated Exocytosis of Lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Castro-Gomes, T.; Corrotte, M.; Tam, C.; Andrews, N.W. Plasma Membrane Repair Is Regulated Extracellularly by Proteases Released from Lysosomes. PLoS ONE 2016, 11, e0152583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, S.M. Axonal Transport and Maturation of Lysosomes. Curr. Opin. Neurobiol. 2018, 51, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Huynh, C.; Roth, D.; Ward, D.M.; Kaplan, J.; Andrews, N.W. Defective Lysosomal Exocytosis and Plasma Membrane Repair in Chediak-Higashi/Beige Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 16795–16800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayó-Puxan, N.; Terrasso, A.P.; Creyssels, S.; Simão, D.; Begon-Pescia, C.; Lavigne, M.; Salinas, S.; Bernex, F.; Bosch, A.; Kalatzis, V.; et al. Lysosomal and Network Alterations in Human Mucopolysaccharidosis Type VII IPSC-Derived Neurons. Sci. Rep. 2018, 8, 16644. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrián, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of Presynaptic Neurotransmission by Macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Vantaggiato, C.; Orso, G.; Guarato, G.; Brivio, F.; Napoli, B.; Panzeri, E.; Masotti, S.; Maria Santorelli, F.; Lamprou, M.; Gumeni, S.; et al. Rescue of Lysosomal Function as Therapeutic Strategy for SPG15 Hereditary Spastic Paraplegia. Brain 2022, awac308. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben-Zvi, H.; Rabinski, T.; Ofir, R.; Cohen, S.; Vatine, G.D. PLEKHM2 Loss of Function Impairs the Activity of iPSC-Derived Neurons via Regulation of Autophagic Flux. Int. J. Mol. Sci. 2022, 23, 16092. https://doi.org/10.3390/ijms232416092
Ben-Zvi H, Rabinski T, Ofir R, Cohen S, Vatine GD. PLEKHM2 Loss of Function Impairs the Activity of iPSC-Derived Neurons via Regulation of Autophagic Flux. International Journal of Molecular Sciences. 2022; 23(24):16092. https://doi.org/10.3390/ijms232416092
Chicago/Turabian StyleBen-Zvi, Hadas, Tatiana Rabinski, Rivka Ofir, Smadar Cohen, and Gad D. Vatine. 2022. "PLEKHM2 Loss of Function Impairs the Activity of iPSC-Derived Neurons via Regulation of Autophagic Flux" International Journal of Molecular Sciences 23, no. 24: 16092. https://doi.org/10.3390/ijms232416092