
The Cell-Specific Role of SHP2 in Regulating Bone Homeostasis and Regeneration Niches



Abstract
:1. Introduction
2. Somatic SHP2 Mutation and Bone Diseases
3. SHP2 in Osteoblast Lineage Cells
3.1. Mesenchymal Stem Cells
3.2. Osteoblasts
3.3. Osteoclast Progenitors/Osteoclasts
3.4. Chondrocytes
4. SHP2 in Bone Niche Cells
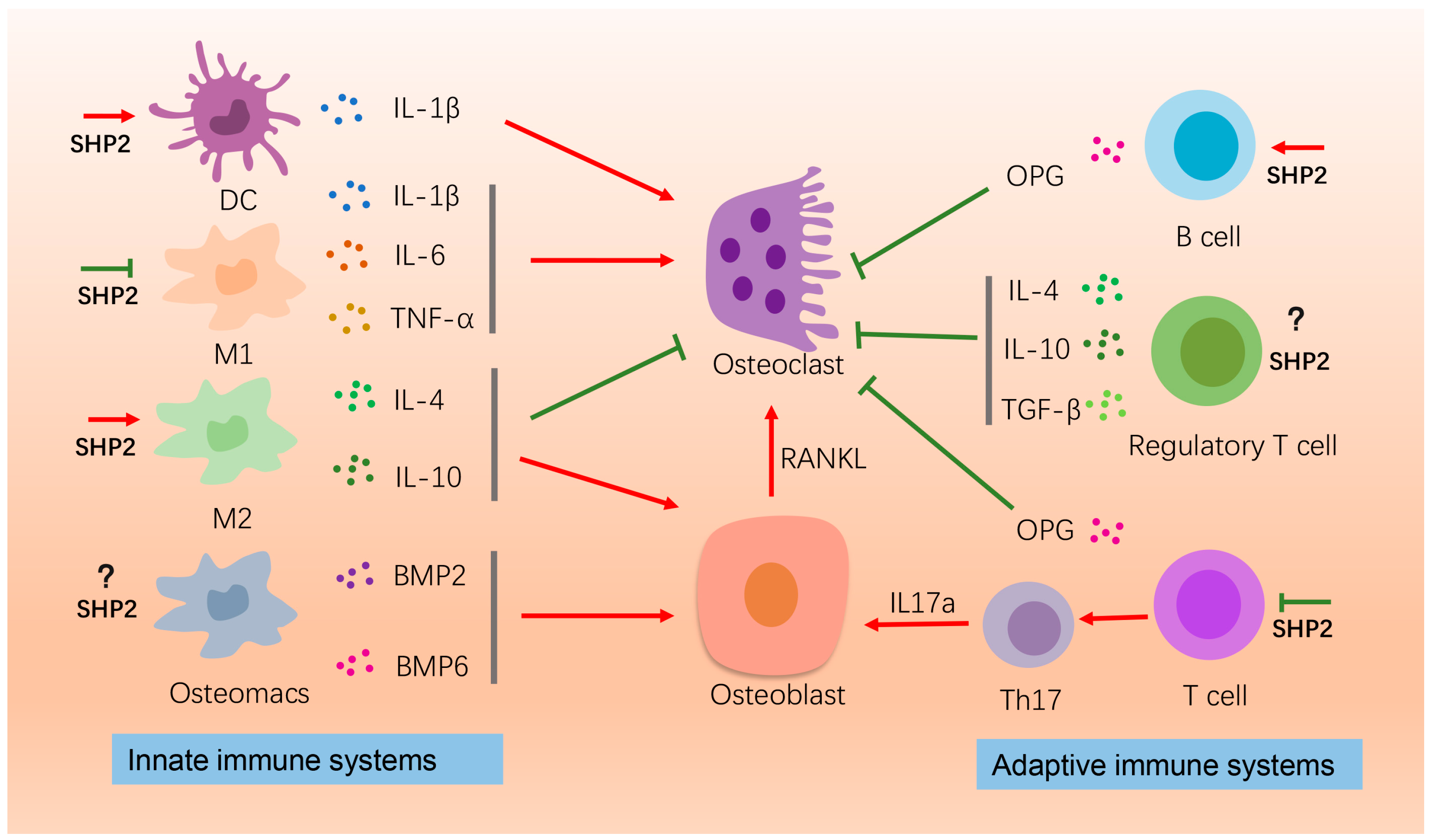
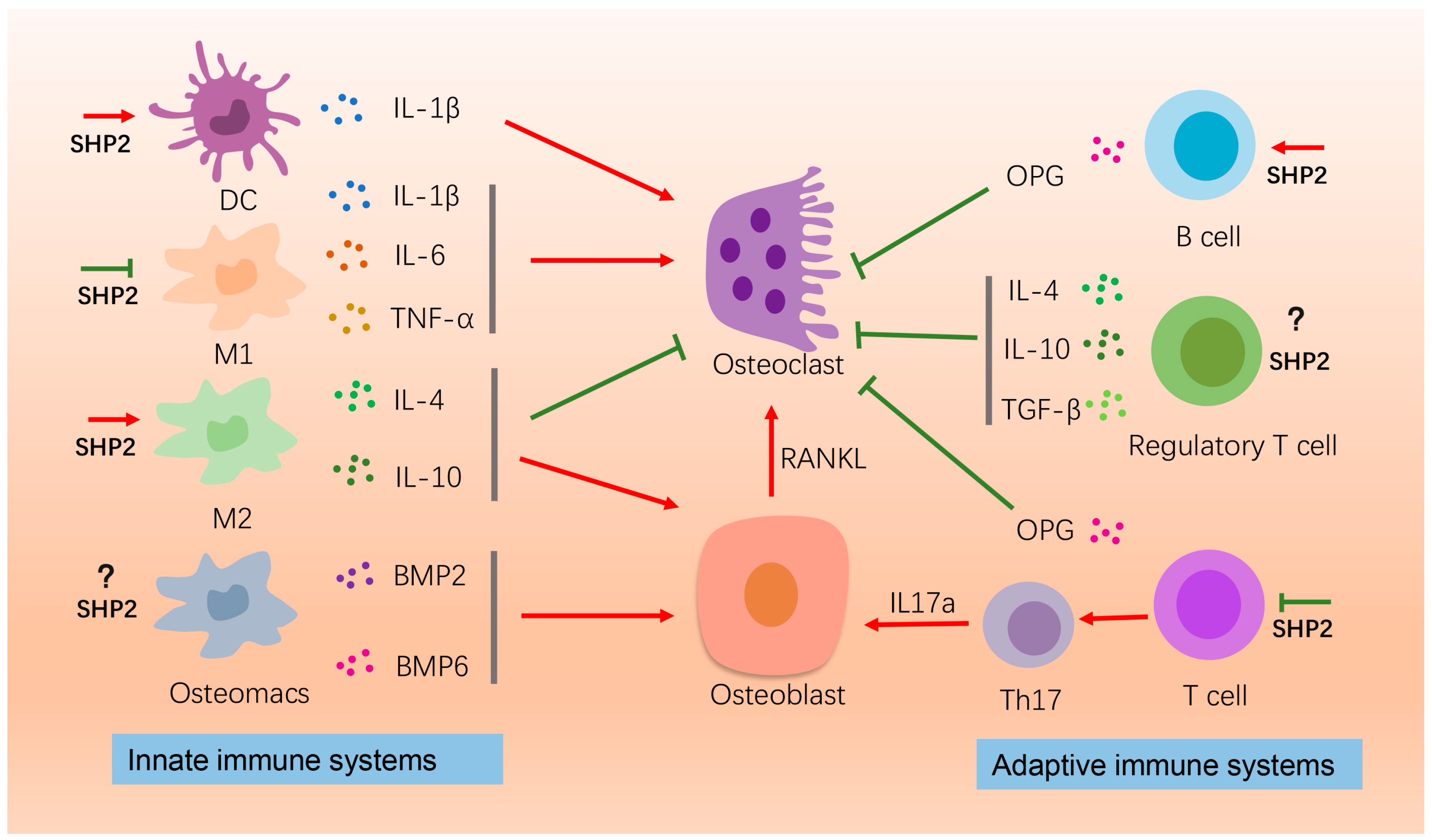
4.1. Immune Cells
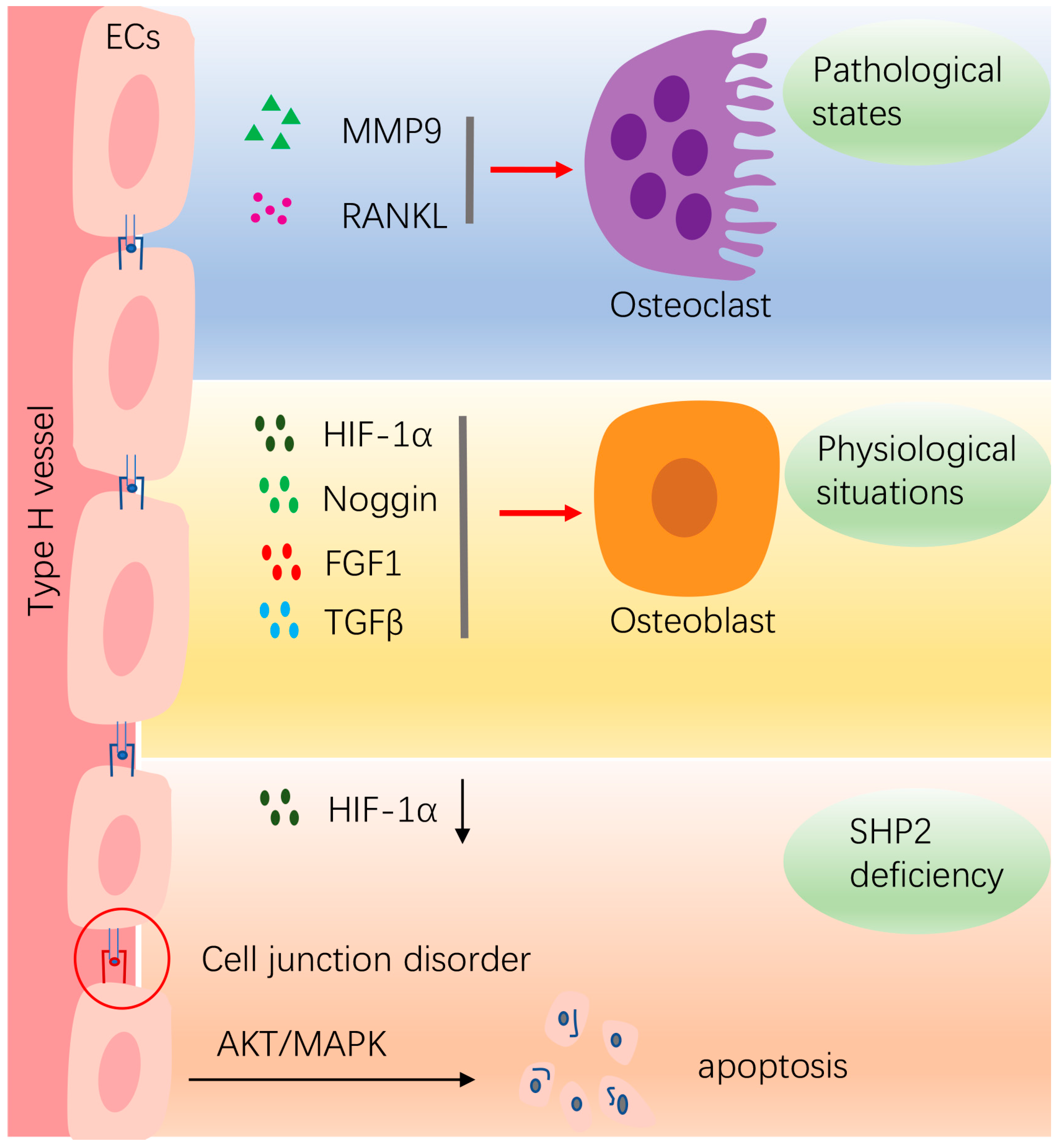
4.2. Vasculature Endothelial Cells
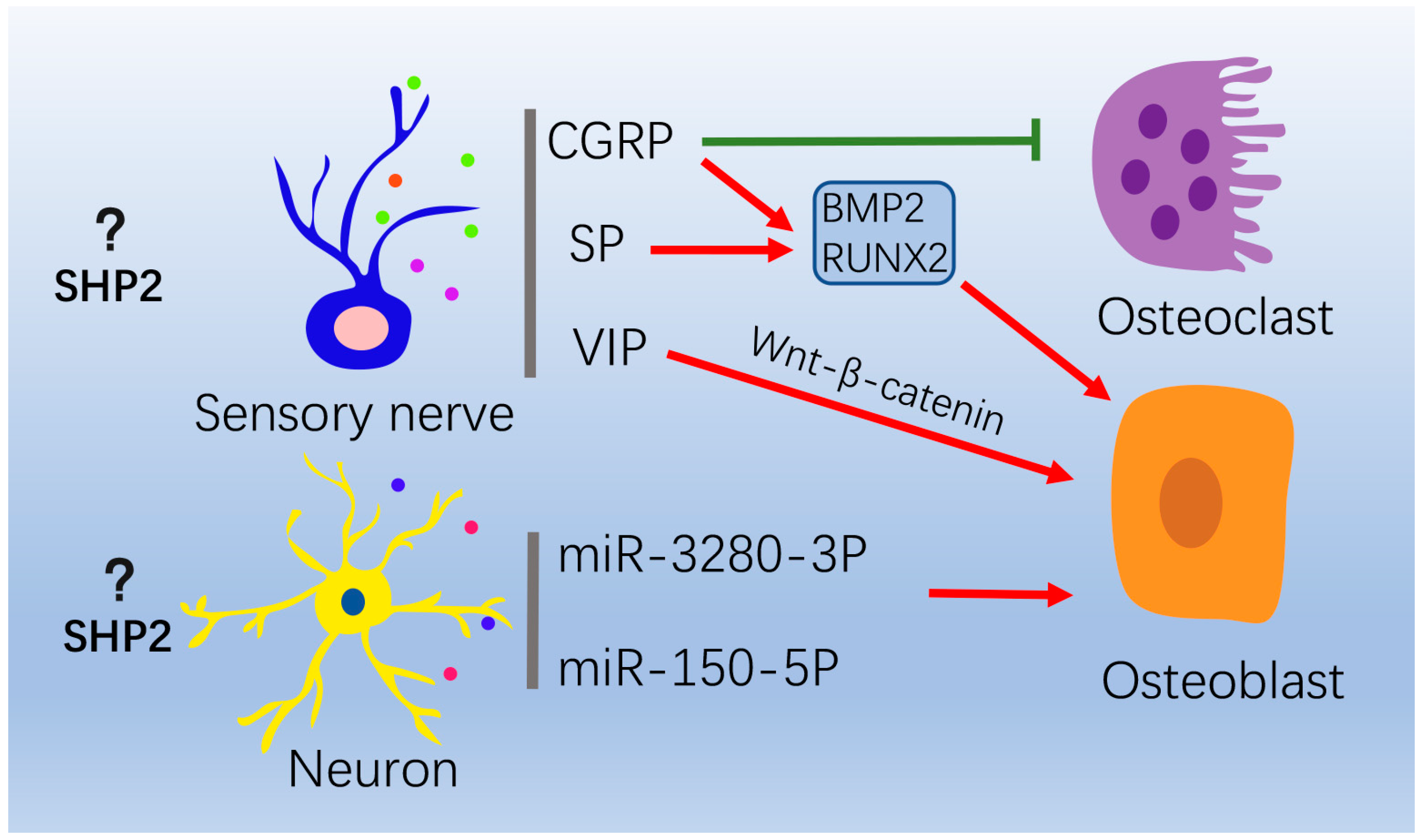
4.3. Nervous System
5. Prospects of SHP2 Agonists and Inhibitors in Treating Bone-Related Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sommerfeldt, D.W.; Rubin, C.T. Biology of bone and how it orchestrates the form and function of the skeleton. Eur. Spine J. 2001, 10, S86–S95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salhotra, A.; Shah, H.N.; Levi, B.; Longaker, M.T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Biol. 2020, 21, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, D.; Martins-Cruz, C.; Oliveira, M.B.; Mano, J.F. Bone physiology as inspiration for tissue regenerative therapies. Biomaterials 2018, 185, 240–275. [Google Scholar] [CrossRef]
- Qin, Q.; Lee, S.; Patel, N.; Walden, K.; Gomez-Salazar, M.; Levi, B.; James, A.W. Neurovascular coupling in bone regeneration. Exp. Mol. Med. 2022, 54, 1844–1849. [Google Scholar] [CrossRef]
- Kamiya, N.; Kim, H.K.; King, P.D. Regulation of bone and skeletal development by the SHP-2 protein tyrosine phosphatase. Bone 2014, 69, 55–60. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, W.; Zhao, Q.; Chen, J.; Wang, T.; Ji, J. The role of the protein tyrosine phosphatase SHP2 in ossification. Dev. Dyn. 2021, 251, 748–758. [Google Scholar] [CrossRef]
- Yang, W.; Wang, J.; Moore, D.C.; Liang, H.; Dooner, M.; Wu, Q.; Terek, R.; Chen, Q.; Ehrlich, M.G.; Quesenberry, P.J.; et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 2013, 499, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.; Kalaitzidis, D.; Neel, B.G. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008, 27, 179–192. [Google Scholar] [CrossRef]
- Yu, Z.-H.; Zhang, R.-Y.; Walls, C.D.; Chen, L.; Zhang, S.; Wu, L.; Liu, S.; Zhang, Z.-Y. Molecular Basis of Gain-of-Function LEOPARD Syndrome-Associated SHP2 Mutations. Biochemistry 2014, 53, 4136–4151. [Google Scholar] [CrossRef]
- Dong, L.; Han, D.; Meng, X.; Xu, M.; Zheng, C.; Xia, Q. Activating Mutation of SHP2 Establishes a Tumorigenic Phonotype Through Cell-Autonomous and Non-Cell-Autonomous Mechanisms. Front. Cell Dev. Biol. 2021, 9, 630712. [Google Scholar] [CrossRef]
- Bowen, M.; Boyden, E.D.; Holm, I.; Campos-Xavier, B.; Bonafé, L.; Superti-Furga, A.; Ikegawa, S.; Cormier-Daire, V.; Bovee, J.; Pansuriya, T.C.; et al. Loss-of-Function Mutations in PTPN11 Cause Metachondromatosis, but Not Ollier Disease or Maffucci Syndrome. PLoS Genet. 2011, 7, e1002050. [Google Scholar] [CrossRef] [PubMed]
- Tajan, M.; Serra, A.D.R.; Valet, P.; Edouard, T.; Yart, A. SHP2 sails from physiology to pathology. Eur. J. Med. Genet. 2015, 58, 509–525. [Google Scholar] [CrossRef]
- Serra-Nédélec, A.D.R.; Edouard, T.; Tréguer, K.; Tajan, M.; Araki, T.; Dance, M.; Mus, M.; Montagner, A.; Tauber, M.; Salles, J.-P.; et al. Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc. Natl. Acad. Sci. USA 2012, 109, 4257–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, M.E.; Ayturk, U.M.; Kurek, K.C.; Yang, W.; Warman, M.L. SHP2 Regulates Chondrocyte Terminal Differentiation, Growth Plate Architecture and Skeletal Cell Fates. PLoS Genet. 2014, 10, e1004364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Feng, G.-S.; Chen, D.; King, P.D.; Kamiya, N. Targeted Disruption of Shp2 in Chondrocytes Leads to Metachondromatosis With Multiple Cartilaginous Protrusions. J. Bone Miner. Res. 2013, 29, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Huang, J.; Moore, D.C.; Song, Y.; Ehrlich, M.G.; Yang, W. SHP2 regulates intramembranous ossification by modifying the TGFβ and BMP2 signaling pathway. Bone 2019, 120, 327–335. [Google Scholar] [CrossRef]
- Wang, L.; Yang, H.; Huang, J.; Pei, S.; Feng, J.Q.; Jing, D.; Zhao, H.; Kronenberg, H.M.; Moore, D.C.; Yang, W. Targeted Ptpn11 deletion in mice reveals the essential role of SHP2 in osteoblast differentiation and skeletal homeostasis. Bone Res. 2021, 9, 6. [Google Scholar] [CrossRef]
- Guo, W.; Liu, W.; Chen, Z.; Gu, Y.; Peng, S.; Shen, L.; Shen, Y.; Wang, X.; Feng, G.-S.; Sun, Y.; et al. Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat. Commun. 2017, 8, 2168. [Google Scholar] [CrossRef] [Green Version]
- Heun, Y.; Pogoda, K.; Anton, M.; Pircher, J.; Pfeifer, A.; Woernle, M.; Ribeiro, A.; Kameritsch, P.; Mykhaylyk, O.; Plank, C.; et al. HIF-1α Dependent Wound Healing Angiogenesis In Vivo Can Be Controlled by Site-Specific Lentiviral Magnetic Targeting of SHP-2. Mol. Ther. 2017, 25, 1616–1627. [Google Scholar] [CrossRef]
- Nakamura, T.; Gulick, J.; Colbert, M.C.; Robbins, J. Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc. Natl. Acad. Sci. USA 2009, 106, 11270–11275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Burgt, I. Noonan syndrome. Orphanet J. Rare Dis. 2007, 2, 4. [Google Scholar] [CrossRef]
- Yart, A.; Edouard, T. Noonan syndrome: An update on growth and development. Curr. Opin. Endocrinol. Diabetes 2018, 25, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkozy, A.; Digilio, M.C.; Dallapiccola, B. Leopard syndrome. Orphanet J. Rare Dis. 2008, 3, 13. [Google Scholar] [CrossRef] [Green Version]
- Aboh, I.V.; Chisci, G.; Gennaro, P.; Gabriele, G.; Cascino, F.; Ginori, A.; Giovannetti, F.; Iannetti, G. LEOPARD Syndrome: Maxillofacial care. J. Craniofacial Surg. 2014, 25, 1094–1095. [Google Scholar] [CrossRef] [PubMed]
- Dzemeshkevich, S.; Frolova, J.; Betekhtin, M.; Shapieva, A.; Rizun, L. The case of 17-year-old male with LEOPARD syndrome. J. Cardiol. Cases 2012, 7, e37–e41. [Google Scholar] [CrossRef] [Green Version]
- Fisher, T.J.; Williams, N.; Morris, L.; Cundy, P.J. Metachondromatosis: More than just multiple osteochondromas. J. Child. Orthop. 2013, 7, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Jamshidi, K.; Shooshtarizadeh, T.; Bahrabadi, M. Chondrosarcoma in Metachondromatosis: A Rare Case Report. Acta Med. Iran. 2017, 55, 793–799. [Google Scholar]
- Tartaglia, M.; Gelb, B.D. Germ-line and somatic PTPN11 mutations in human disease. Eur. J. Med. Genet. 2005, 48, 81–96. [Google Scholar] [CrossRef]
- Hasle, H. Malignant Diseases in Noonan Syndrome and Related Disorders. Horm. Res. Paediatr. 2009, 72, 8–14. [Google Scholar] [CrossRef]
- Moore, E.R.; Chen, J.C.; Jacobs, C.R. Prx1-Expressing Progenitor Primary Cilia Mediate Bone Formation in response to Mechanical Loading in Mice. Stem Cells Int. 2019, 2019, 3094154. [Google Scholar] [CrossRef]
- de Lageneste, O.D.; Julien, A.; Abou-Khalil, R.; Frangi, G.; Carvalho, C.; Cagnard, N.; Cordier, C.; Conway, S.J.; Colnot, C. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat. Commun. 2018, 9, 773. [Google Scholar] [CrossRef] [Green Version]
- Zuo, C.; Wang, L.; Kamalesh, R.M.; Bowen, M.E.; Moore, D.C.; Dooner, M.S.; Reginato, A.M.; Wu, Q.; Schorl, C.; Song, Y.; et al. SHP2 regulates skeletal cell fate by modifying SOX9 expression and transcriptional activity. Bone Res. 2018, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Mohan, A.; Moore, D.C.; Lin, L.; Zhou, F.L.; Cao, J.; Wu, Q.; Qin, Y.; Reginato, A.M.; Ehrlich, M.G.; et al. SHP2 regulates osteoclastogenesis by promoting preosteoclast fusion. FASEB J. 2015, 29, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.W.; Aruwajoye, O.; Sucato, D.; Richards, B.S.; Feng, G.-S.; Chen, D.; King, P.D.; Kamiya, N. Induction of SHP2 Deficiency in Chondrocytes Causes Severe Scoliosis and Kyphosis in Mice. Spine 2013, 38, E1307–E1312. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, N.; Shen, J.; Noda, K.; Kitami, M.; Feng, G.-S.; Chen, D.; Komatsu, Y. SHP2-Deficiency in Chondrocytes Deforms Orofacial Cartilage and Ciliogenesis in Mice. J. Bone Miner. Res. 2015, 30, 2028–2032. [Google Scholar] [CrossRef]
- Wang, L.J.; Huang, J.H.; Moore, U.C.; Zuo, C.L.; Wu, Q.; Xie, L.Q.; Von Der Mark, K.; Yuan, X.; Chen, D.; Warman, M.L.; et al. SHP2 Regulates the Osteogenic Fate of Growth Plate Hypertrophic Chondrocytes. Sci. Rep. 2017, 7, 12699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, F.; Liu, Q.; Zhu, Y.; Fan, Z.; Chen, W.; Liu, S.; Li, X.; Guo, W.; Feng, G.-S.; Yu, H.; et al. Targeting chondrocytes for arresting bony fusion in ankylosing spondylitis. Nat. Commun. 2021, 12, 6540. [Google Scholar] [CrossRef]
- Miah, S.M.S.; Jayasuriya, C.T.; Salter, A.; Reilly, E.C.; Fugere, C.; Yang, W.; Chen, Q.; Brossay, L. Ptpn11 Deletion in CD4+ Cells Does Not Affect T Cell Development and Functions but Causes Cartilage Tumors in a T Cell-Independent Manner. Front. Immunol. 2017, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Liu, S.; Jiang, S.; Li, Z.; Mo, X.; Ruan, H.; Zou, G.-M.; Fan, C. The use of SHP-2 gene transduced bone marrow mesenchymal stem cells to promote osteogenic differentiation and bone defect repair in rat. J. Biomed. Mater. Res. Part A 2016, 104, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.-P.; Deng, M.; Chen, L.-Y.; Fang, N.; Du, C.; Chen, L.; Zou, Y.; Dai, J.; Zhu, M.; Wang, W.; et al. Shp2 regulates chlorogenic acid-induced proliferation and adipogenic differentiation of bone marrow-derived mesenchymal stem cells in adipogenesis. Mol. Med. Rep. 2015, 11, 4489–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.P.; Lin, S.J.; Wan, W.B.; Zuo, H.L.; Yao, F.F.; Ruan, H.B.; Xu, J.; Song, W.; Zhou, Y.C.; Wen, S.Y.; et al. Chlorogenic Acid Prevents Osteoporosis by Shp2/PI3K/Akt Pathway in Ovariectomized Rats. PLoS ONE 2016, 11, e0166751. [Google Scholar] [CrossRef] [Green Version]
- Roubelakis, M.G.; Tsaknakis, G.; Lyu, F.-J.; Trohatou, O.; Zannettino, A.C.W.; Watt, S.M. P0-Related Protein Accelerates Human Mesenchymal Stromal Cell Migration by Modulating VLA-5 Interactions with Fibronectin. Cells 2020, 9, 1100. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jing, H.; Kou, X.; Chen, C.; Liu, D.; Jin, Y.; Lu, L.; Shi, S. PD-1 is required to maintain stem cell properties in human dental pulp stem cells. Cell Death Differ. 2018, 25, 1350–1360. [Google Scholar] [CrossRef]
- Lapinski, P.E.; Meyer, M.F.; Feng, G.-S.; Kamiya, N.; King, P.D. Deletion of SHP-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity, and calvarial defects in mice. Dis. Model. Mech. 2013, 6, 1448–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauler, T.; Kamiya, N.; Lapinski, P.E.; Langewisch, E.; Mishina, Y.; Wilkinson, J.E.; Feng, G.-S.; King, P.D. Development of severe skeletal defects in induced SHP-2-deficient adult mice: A model of skeletal malformation in humans with SHP-2 mutations. Dis. Model. Mech. 2011, 4, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Iorio, C.; Yan, K.; Yang, H.; Takeshita, S.; Kang, S.; Neel, B.G.; Yang, W. A ERK/RSK-mediated negative feedback loop regulates M-CSF–evoked PI3K/AKT activation in macrophages. FASEB J. 2018, 32, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Sims, N.A.; Jenkins, B.J.; Quinn, J.M.; Nakamura, A.; Glatt, M.; Gillespie, M.T.; Ernst, M.; Martin, T.J. Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J. Clin. Investig. 2004, 113, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Tao, T.; Luo, D.; Gao, C.; Liu, H.; Lei, Z.; Liu, W.; Zhou, C.; Qi, D.; Deng, Z.; Sun, X.; et al. Src Homology 2 Domain-Containing Protein Tyrosine Phosphatase Promotes Inflammation and Accelerates Osteoarthritis by Activating β-Catenin. Front. Cell Dev. Biol. 2021, 9, 646386. [Google Scholar] [CrossRef]
- Liu, Q.; Zhai, L.; Han, M.; Shi, D.; Sun, Z.; Peng, S.; Wang, M.; Zhang, C.; Gao, J.; Yan, W.; et al. SH2 Domain–Containing Phosphatase 2 Inhibition Attenuates Osteoarthritis by Maintaining Homeostasis of Cartilage Metabolism via the Docking Protein 1/Uridine Phosphorylase 1/Uridine Cascade. Arthritis Rheumatol. 2022, 74, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Kang, L.; Li, C.; Chen, S.; Wang, Q.; Yang, J.; Long, Y.; Li, J.; Zhao, K.; Xu, W.; et al. A self-powered implantable and bioresorbable electrostimulation device for biofeedback bone fracture healing. Proc. Natl. Acad. Sci. USA 2021, 118, e2100772118. [Google Scholar] [CrossRef]
- Walsh, M.C.; Takegahara, N.; Kim, H.; Choi, Y. Updating osteoimmunology: Regulation of bone cells by innate and adaptive immunity. Nat. Rev. Rheumatol. 2018, 14, 146–156. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X.; Liu, J.; Zheng, Y.; Wu, Y.; Yang, W.; Yi, Y.; Liu, J.; Wang, J. d-mannose attenuates lipopolysaccharide-induced osteolysis via CPT1A-Mediated lipid metabolic regulation in macrophages. Biochem. Biophys. Res. Commun. 2021, 583, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Graney, P.L.; Roohani-Esfahani, S.I.; Zreiqat, H.; Spiller, K.L. In vitro response of macrophages to ceramic scaffolds used for bone regeneration. J. R. Soc. Interface 2016, 13, 20160346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotchkiss, K.M.; Reddy, G.B.; Hyzy, S.L.; Schwartz, Z.; Boyan, B.D.; Olivares-Navarrete, R. Titanium surface characteristics, including topography and wettability, alter macrophage activation. Acta Biomater. 2015, 31, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Zhao, Y.; Zhang, Y.; Ruan, Z. The Macrophage Polarization Regulates MSC Osteoblast Differentiation in vitro. Ann. Clin. Lab. Sci. 2016, 46, 65–71. [Google Scholar] [PubMed]
- von Kaeppler, E.P.; Wang, Q.; Raghu, H.; Bloom, M.S.; Wong, H.; Robinson, W.H. Interleukin 4 promotes anti-inflammatory macrophages that clear cartilage debris and inhibits osteoclast development to protect against osteoarthritis. Clin. Immunol. 2021, 229, 108784. [Google Scholar] [CrossRef]
- Xiao, P.; Guo, Y.; Zhang, H.; Zhang, X.; Cheng, H.; Cao, Q.; Ke, Y. Myeloid-restricted ablation of Shp2 restrains melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-γ production in tumor microenvironment. Oncogene 2018, 37, 5088–5100. [Google Scholar] [CrossRef]
- Wang, Y.; Mohseni, M.; Grauel, A.; Diez, J.E.; Guan, W.; Liang, S.; Choi, J.E.; Pu, M.; Chen, D.; Laszewski, T.; et al. SHP2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci. Rep. 2021, 11, 1399. [Google Scholar] [CrossRef]
- Batoon, L.; Millard, S.; Raggatt, L.J.; Pettit, A.R. Osteomacs and Bone Regeneration. Curr. Osteoporos. Rep. 2017, 15, 385–395. [Google Scholar] [CrossRef]
- Batoon, L.; Millard, S.M.; Wullschleger, M.E.; Preda, C.; Wu, A.C.-K.; Kaur, S.; Tseng, H.-W.; Hume, D.A.; Levesque, J.-P.; Raggatt, L.J.; et al. CD169+ macrophages are critical for osteoblast maintenance and promote intramembranous and endochondral ossification during bone repair. Biomaterials 2019, 196, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibáñez, L.; Abou-Ezzi, G.; Ciucci, T.; Amiot, V.; Belaïd, N.; Obino, D.; Mansour, A.; Rouleau, M.; Wakkach, A.; Blin-Wakkach, C. Inflammatory Osteoclasts Prime TNFα-Producing CD4+T Cells and Express CX3CR1. J. Bone Miner. Res. 2016, 31, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Y.; Yang, Y.; Liu, Y.; Tian, Q.; Liu, P.; Ding, Z.; Cheng, H.; Zhang, X.; Ke, Y. Tyrosine phosphatase Shp2 regulates p115RhoGEF/Rho-dependent dendritic cell migration. Cell. Mol. Immunol. 2020, 18, 755–757. [Google Scholar] [CrossRef]
- Weitzmann, M.N.; Ofotokun, I. Physiological and pathophysiological bone turnover—Role of the immune system. Nat. Rev. Endocrinol. 2016, 12, 518–532. [Google Scholar] [CrossRef]
- Li, Y.; Toraldo, G.; Li, A.; Yang, X.; Zhang, H.; Qian, W.-P.; Weitzmann, M.N. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 2007, 109, 3839–3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitzmann, M.N. Bone and the Immune System. Toxicol. Pathol. 2017, 45, 911–924. [Google Scholar] [CrossRef]
- Friederichs, K.; Schmitz, J.; Weissenbach, M.; Heinrich, P.C.; Schaper, F. Interleukin-6-induced proliferation of pre-B cells mediated by receptor complexes lacking the SHP2/SOCS3 recruitment sites revisited. JBIC J. Biol. Inorg. Chem. 2001, 268, 6401–6407. [Google Scholar] [CrossRef]
- Tamir, I.; Porto, J.M.D.; Cambier, J.C. Cytoplasmic protein tyrosine phosphatases SHP-1 and SHP-2: Regulators of B cell signal transduction. Curr. Opin. Immunol. 2000, 12, 307–315. [Google Scholar] [CrossRef]
- Bozec, A.; Zaiss, M.M. T Regulatory Cells in Bone Remodelling. Curr. Osteoporos. Rep. 2017, 15, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, E.; Partap, S.; Azevedo, M.M.; Jell, G.; Stevens, M.M.; O’Brien, F.J. Hypoxia-mimicking bioactive glass/collagen glycosaminoglycan composite scaffolds to enhance angiogenesis and bone repair. Biomaterials 2015, 52, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-Y.; Lin, R.-B.; Huang, S.-H.; Liang, Y.-J.; Li, X.; Zhang, S.-E.; Ouyang, D.-Q.; Li, K.; Zheng, G.-S.; Liao, G.-Q. PDGF-BB exhibited therapeutic effects on rat model of bisphosphonate-related osteonecrosis of the jaw by enhancing angiogenesis and osteogenesis. Bone 2019, 144, 115117. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328, Erratum in Nature 2014, 513, 574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pan, J.; Jing, W. Motivating role of type H vessels in bone regeneration. Cell Prolif. 2020, 53, e12874. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, S.; Li, Y.; Crane, J.L. Type H blood vessels in bone modeling and remodeling. Theranostics 2020, 10, 426–436. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, H.; Cai, D.; Zeng, C.; Lai, P.; Shao, Y.; Fang, H.; Li, D.; Ouyang, J.; Zhao, C.; et al. Positive-Feedback Regulation of Subchondral H-Type Vessel Formation by Chondrocyte Promotes Osteoarthritis Development in Mice. J. Bone Miner. Res. 2018, 33, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, J.; Qi, T.; Huang, Y.; Lu, Y.; Zhan, T.; Gong, H.; Zhu, Z.; Shi, Y.; Zhou, J.; et al. SHP2 protects endothelial cell barrier through suppressing VE-cadherin internalization regulated by MET-ARF1. FASEB J. 2018, 33, 1124–1137. [Google Scholar] [CrossRef]
- Rieck, S.; Heun, Y.; Heidsieck, A.; Mykhaylyk, O.; Pfeifer, A.; Gleich, B.; Mannell, H.; Wenzel, D. Local anti-angiogenic therapy by magnet-assisted downregulation of SHP2 phosphatase. J. Control. Release 2019, 305, 155–164. [Google Scholar] [CrossRef]
- Wang, Y.; Salvucci, O.; Ohnuki, H.; Tran, A.D.; Ha, T.; Feng, J.; DiPrima, M.; Kwak, H.; Wang, D.; Yu, Y.; et al. Targeting the SHP2 phosphatase promotes vascular damage and inhibition of tumor growth. EMBO Mol. Med. 2021, 13, e14089. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Guo, C.; Ye, Q.; Shi, Y.; Sun, Y.; Zhang, J.; Huang, J.; Huang, Y.; Zeng, C.; Zhang, X.; et al. Endothelial deletion of SHP2 suppresses tumor angiogenesis and promotes vascular normalization. Nat. Commun. 2021, 12, 6310. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Xie, J.; Cai, Z.; Liu, X.; Wen, J.; Cui, Z.-K.; Zhao, R.; Zhou, X.; Chen, J.; Mao, X.; et al. Damaged brain accelerates bone healing by releasing small extracellular vesicles that target osteoprogenitors. Nat. Commun. 2021, 12, 6043. [Google Scholar] [CrossRef] [PubMed]
- Idelevich, A.; Baron, R. Brain to bone: What is the contribution of the brain to skeletal homeostasis? Bone 2018, 115, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Elefteriou, F.; Takeda, S.; Ebihara, K.; Magre, J.; Patano, N.; Kim, C.A.; Ogawa, Y.; Liu, X.; Ware, S.M.; Craigen, W.J.; et al. Serum leptin level is a regulator of bone mass. Proc. Natl. Acad. Sci. USA 2004, 101, 3258–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offley, S.C.; Guo, T.Z.; Wei, T.; Clark, J.D.; Vogel, H.; Lindsey, D.P.; Jacobs, C.R.; Yao, W.; Lane, N.E.; Kingery, W.S. Capsaicin-Sensitive Sensory Neurons Contribute to the Maintenance of Trabecular Bone Integrity. J. Bone Miner. Res. 2004, 20, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Matsusue, Y. Neuronal regulation of bone metabolism and anabolism: Calcitonin gene-related peptide-, substance P-, and tyrosine hydroxylase-containing nerves and the bone. Microsc. Res. Tech. 2002, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Orita, S.; Ohtori, S.; Koshi, T.; Yamashita, M.; Yamauchi, K.; Inoue, G.; Suzuki, M.; Eguchi, Y.; Kamoda, H.; Arai, G.; et al. The Effects of Risedronate and Exercise on Osteoporotic Lumbar Rat Vertebrae and Their Sensory Innervation. Spine 2010, 35, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; Chen, X.; Li, Y.; Mi, J.; Qin, L. The Effects of Calcitonin Gene-Related Peptide on Bone Homeostasis and Regeneration. Curr. Osteoporos. Rep. 2020, 18, 621–632. [Google Scholar] [CrossRef]
- Li, J.; Kreicbergs, A.; Bergström, J.; Stark, A.; Ahmed, M. Site-specific CGRP innervation coincides with bone formation during fracture healing and modeling: A study in rat angulated tibia. J. Orthop. Res. 2007, 25, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Park, B.-W.; Kim, J.-R.; Lee, J.-H.; Byun, J.-H. Expression of nerve growth factor and vascular endothelial growth factor in the inferior alveolar nerve after distraction osteogenesis. Int. J. Oral Maxillofac. Surg. 2006, 35, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Tuzmen, C.; Campbell, P.G. Crosstalk between neuropeptides SP and CGRP in regulation of BMP2-induced bone differentiation. Connect. Tissue Res. 2018, 59, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Feng, L.; Zhu, M.-L.; Yang, Z.-M.; Wu, T.-Y.; Xu, J.; Liu, Y.; Lin, W.-P.; Lo, J.H.T.; Zhang, J.-F.; et al. Vasoactive Intestinal Peptide Stimulates Bone Marrow-Mesenchymal Stem Cells Osteogenesis Differentiation by Activating Wnt/β-Catenin Signaling Pathway and Promotes Rat Skull Defect Repair. Stem Cells Dev. 2020, 29, 655–666. [Google Scholar] [CrossRef]
- Ke, Y.; Zhang, E.E.; Hagihara, K.; Wu, D.; Pang, Y.; Klein, R.; Curran, T.; Ranscht, B.; Feng, G.-S. Deletion of Shp2 in the Brain Leads to Defective Proliferation and Differentiation in Neural Stem Cells and Early Postnatal Lethality. Mol. Cell. Biol. 2007, 27, 6706–6717. [Google Scholar] [CrossRef] [Green Version]
- Hagihara, K.; Zhang, E.; Ke, Y.-H.; Liu, G.; Liu, J.-J.; Rao, Y.; Feng, G.-S. Shp2 acts downstream of SDF-1α/CXCR4 in guiding granule cell migration during cerebellar development. Dev. Biol. 2009, 334, 276–284. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Zhang, S.S.; Meng, Q.; Li, S.; Zhu, H.H.; Raquil, M.-A.; Alderson, N.; Zhang, H.; Wu, J.; Rui, L.; et al. Shp2 Controls Female Body Weight and Energy Balance by Integrating Leptin and Estrogen Signals. Mol. Cell. Biol. 2012, 32, 1867–1878. [Google Scholar] [CrossRef] [Green Version]
- Krajewska, M.; Banares, S.; Zhang, E.E.; Huang, X.; Scadeng, M.; Jhala, U.S.; Feng, G.-S.; Krajewski, S. Development of Diabesity in Mice with Neuronal Deletion of Shp2 Tyrosine Phosphatase. Am. J. Pathol. 2008, 172, 1312–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, M.; Shen, L.; Zhu, Y.; Ma, H.; Liu, B.; Ouyang, L.; Guo, W.; Xu, Q.; Sun, Y. SHP2-mediated mitophagy boosted by lovastatin in neuronal cells alleviates parkinsonism in mice. Signal Transduct. Target. Ther. 2021, 6, 34. [Google Scholar] [CrossRef]
- Xie, L.; Zhu, G.; Shang, J.; Chen, X.; Zhang, C.; Ji, X.; Zhang, Q.; Wei, Y. An overview on the biological activity and anti-cancer mechanism of lovastatin. Cell. Signal. 2021, 87, 110122. [Google Scholar] [CrossRef]
- Garrett, I.; Gutierrez, G.; Rossini, G.; Nyman, J.; McCluskey, B.; Flores, A.; Mundy, G. Locally delivered lovastatin nanoparticles enhance fracture healing in rats. J. Orthop. Res. 2007, 25, 1351–1357. [Google Scholar] [CrossRef]
- Bleedorn, J.A.; Sullivan, R.; Lu, Y.; Nemke, B.; Kalscheur, V.; Markel, M.D. Percutaneous lovastatin accelerates bone healing but is associated with periosseous soft tissue inflammation in a canine tibial osteotomy model. J. Orthop. Res. 2013, 32, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Montoya, C.J.; Jaimes, F.; Higuita, E.A.; Convers-Paez, S.; Estrada, S.; Gutierrez, F.; Amariles, P.; Giraldo, N.; Penaloza, C.; Rugeles, M.T. Antiretroviral effect of lovastatin on HIV-1-infected individuals without highly active antiretroviral therapy (The LIVE study): A phase-II randomized clinical trial. Trials 2009, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Amadasu, E.; Kang, R.; Usmani, A.; Borlongan, C.V. Effects of Lovastatin on Brain Cancer Cells. Cell Transplant. 2022, 31, 9636897221102903. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-N.P.; Lamarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Miao, L.; Lin, H.; Cheng, J.; Li, M.; Zhuo, Z.; He, J. Targeting RAS in neuroblastoma: Is it possible? Pharmacol. Ther. 2022, 236, 108054. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wu, Z.; Zhao, M.; Zhang, R.; Li, M.; Sun, D.; Cheng, H.; Qi, X.; Shen, Y.; Xu, Q.; et al. Allosteric inhibition reveals SHP2-mediated tumor immunosuppression in colon cancer by single-cell transcriptomics. Acta Pharm. Sin. B 2021, 12, 149–166. [Google Scholar] [CrossRef]
- Kano, H.; Ichihara, E.; Watanabe, H.; Nishii, K.; Ando, C.; Nakasuka, T.; Ninomiya, K.; Kato, Y.; Kubo, T.; Rai, K.; et al. SHP2 Inhibition Enhances the Effects of Tyrosine Kinase Inhibitors in Preclinical Models of Treatment-naïve ALK-, ROS1-, or EGFR-altered Non–small Cell Lung Cancer. Mol. Cancer Ther. 2021, 20, 1653–1662. [Google Scholar] [CrossRef]
- Wang, J.; Huang, L.; Huang, Y.; Jiang, Y.; Zhang, L.; Feng, G.; Liu, L. Therapeutic effect of the injectable thermosensitive hydrogel loaded with SHP099 on intervertebral disc degeneration. Life Sci. 2020, 266, 118891. [Google Scholar] [CrossRef]
- Yin, H.; Huang, J.; Cao, X.; Wang, Z.-X.; Cao, J.; Hu, Y.; Luo, J.; Tan, Y.-J.; Chen, T.-H.; Chen, C.-Y.; et al. Inhibition of Src Homology 2 Domain-Containing Protein Tyrosine Phosphatase-2 Facilitates CD31hiEndomucinhi Blood Vessel and Bone Formation in Ovariectomized Mice. Cell. Physiol. Biochem. 2018, 50, 1068–1083. [Google Scholar] [CrossRef]
- LaMarche, M.J.; Acker, M.G.; Argintaru, A.; Bauer, D.; Boisclair, J.; Chan, H.; Chen, C.H.-T.; Chen, Y.-N.P.; Chen, Z.; Deng, Z.; et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 13578–13594. [Google Scholar] [CrossRef]
- Shen, D.; Chen, W.; Zhu, J.; Wu, G.; Shen, R.; Xi, M.; Sun, H. Therapeutic potential of targeting SHP2 in human developmental disorders and cancers. Eur. J. Med. Chem. 2020, 190, 112117. [Google Scholar] [CrossRef] [PubMed]
- Tajan, M.; Pernin-Grandjean, J.; Beton, N.; Gennero, I.; Capilla, F.; Neel, B.G.; Araki, T.; Valet, P.; Tauber, M.; Salles, J.-P.; et al. Noonan syndrome-causing SHP2 mutants impair ERK-dependent chondrocyte differentiation during endochondral bone growth. Hum. Mol. Genet. 2018, 27, 2276–2289. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Target Cell | Phenotype | Mechanism | Related Signaling Pathways | References |
|---|---|---|---|---|
| Prrx1+ mesenchyme stem cells | Skeletal dysplasia; impaired ossification in skull, long bones, ribs, limbs and joint; pectus excavatum and pectus carinatum; endochondral ossification; exostoses | Chondrogenic transcription factors: SOX9, Acan, Col2a1, Col10a1↑ Osteogenic transcription factors: ALP, Col1a1, Ctnnb1, Sp7, RUNX2↓ | TGF-β/SMAD2/3, BMP2/SMAD1/5/8↓ | [17,32,33,34] |
| Bglap+ osteoblasts | Scoliosis, osteoporosis, osteochondromas, enchondromas | Osteogenic transcription factors: RUNX2, Osterix7, Dmp1, Sost↓ | STAT3/RANKL↑ | [18] |
| LysM+ osteoclasts precursors | Age-related osteopetrosis | Osteoclastogenesis transcription factors: Nfatc1↓ | AKT↓, RAS /ERK↓ | [8] |
| CTSK+ osteoclasts | Osteopetrosis, scoliosis, exostoses and enchondromas | Reducing osteoclasts activity | MAPK↓, IHH↑ | [8,35] |
| Col2a1+ chondrocytes | Spinal deformities, scoliosis, kyphosis, lordosis, enchondroma and exostosis | Chondrogenic transcription factors: SOX9, BMP6↑ Osteogenic transcription factors: ALP↓ | IHH↑, MAPK↓, β-catenin↓ | [15,16,36,37] |
| Fsp1+ expressing fibroblasts | Exostosis | Inducing normal cells undergo chondrogenesis by paracrine | Unknow | [15] |
| Col10a-1+ chondrocytes | Bone mineral density reduction | Chondrogenic transcription factors: SOX9↑ Osteogenic transcription factors: Ibsp, RUNX2, Ctnnb1↓ | WNT/β-catenin↓ | [38] |
| CD4+ chondrocytes | Bone fusion and joint stiffness, ankylosing spondylitis, osteoporosis | Chondrogenic transcription factors: Col2a1, Col10a1, Acan, and Pthrp↑ Osteogenic transcription factors: RUNX2, Sp7, Ocn↑ | BMP6/Smad1/5↑, ERK1/2↓ IHH↓ | [39,40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Ye, C.; Zhu, Y.; Wang, J.; Liu, J. The Cell-Specific Role of SHP2 in Regulating Bone Homeostasis and Regeneration Niches. Int. J. Mol. Sci. 2023, 24, 2202. https://doi.org/10.3390/ijms24032202
Zhang J, Ye C, Zhu Y, Wang J, Liu J. The Cell-Specific Role of SHP2 in Regulating Bone Homeostasis and Regeneration Niches. International Journal of Molecular Sciences. 2023; 24(3):2202. https://doi.org/10.3390/ijms24032202
Chicago/Turabian StyleZhang, Jie, Chengxinyue Ye, Yufan Zhu, Jun Wang, and Jin Liu. 2023. "The Cell-Specific Role of SHP2 in Regulating Bone Homeostasis and Regeneration Niches" International Journal of Molecular Sciences 24, no. 3: 2202. https://doi.org/10.3390/ijms24032202
APA StyleZhang, J., Ye, C., Zhu, Y., Wang, J., & Liu, J. (2023). The Cell-Specific Role of SHP2 in Regulating Bone Homeostasis and Regeneration Niches. International Journal of Molecular Sciences, 24(3), 2202. https://doi.org/10.3390/ijms24032202