
Neuromuscular and Neuromuscular Junction Manifestations of the PURA-NDD: A Systematic Review of the Reported Symptoms and Potential Treatment Options


Abstract
:1. Introduction
1.1. Molecular Function of PURA and Its Role in the Central Nervous System and Other Tissues
1.2. PURA’s Role in the Muscle
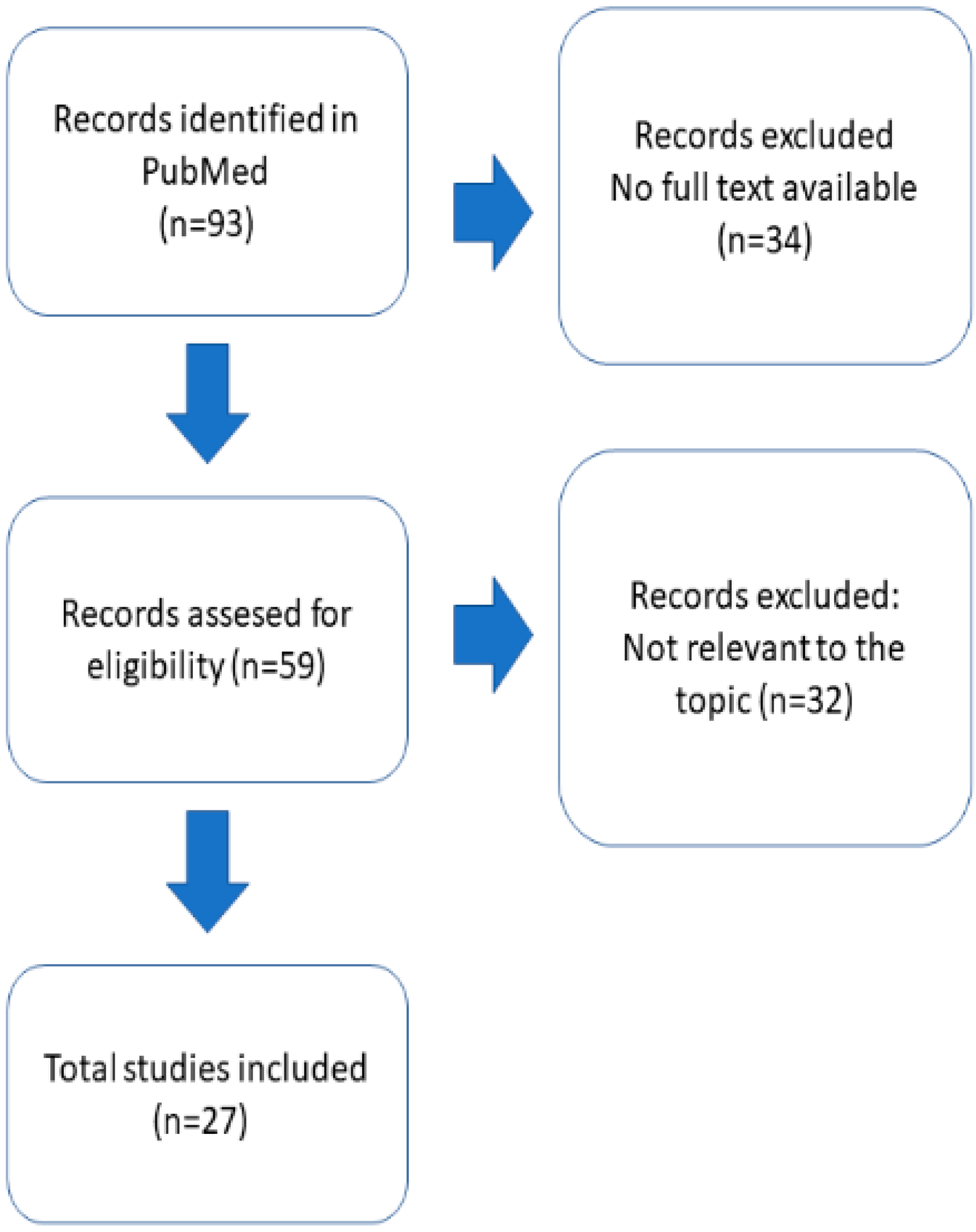
2. Materials and Methods
3. Results and Discussion
3.1. Motor and Neuromuscular Symptoms in PURA Syndrome
3.2. Electrophysiological and Morphological Studies
3.3. Limitations
3.4. Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, E.M.; Daniel, D.C.; Gordon, J. The pur protein family: Genetic and structural features in development and disease. J. Cell Physiol. 2013, 228, 930–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molitor, L.; Bacher, S.; Burczyk, S.; Niessing, D. The Molecular Function of PURA and Its Implications in Neurological Diseases. Front. Genet. 2021, 12, 638217. [Google Scholar] [CrossRef] [PubMed]
- Daniel, D.C.; Johnson, E.M. PURA, the gene encoding Pur-alpha, member of an ancient nucleic acid-binding protein family with mammalian neurological functions. Gene 2018, 643, 133–143. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Johnson, E.M.; Khalili, K. Multiple roles for Puralpha in cellular and viral regulation. Cell Cycle 2009, 8, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Sueblinvong, V.; Raman, J.; Jeevanandam, V.; Gupta, M.P. Single-stranded DNA-binding proteins PURalpha and PURbeta bind to a purine-rich negative regulatory element of the alpha-myosin heavy chain gene and control transcriptional and translational regulation of the gene expression. Implications in the repression of alpha-myosin heavy chain during heart failure. J. Biol. Chem. 2003, 278, 44935–44948. [Google Scholar] [CrossRef] [Green Version]
- Lalani, S.R.; Zhang, J.; Schaaf, C.P.; Brown, C.W.; Magoulas, P.; Tsai, A.C.; El-Gharbawy, A.; Wierenga, K.J.; Bartholomew, D.; Fong, C.T.; et al. Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am. J. Hum. Genet. 2014, 95, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Hosoki, K.; Ohta, T.; Natsume, J.; Imai, S.; Okumura, A.; Matsui, T.; Harada, N.; Bacino, C.A.; Scaglia, F.; Jones, J.Y.; et al. Clinical phenotype and candidate genes for the 5q31.3 microdeletion syndrome. Am. J. Med. Genet. A 2012, 158A, 1891–1896. [Google Scholar] [CrossRef]
- Dai, W.; Sun, Y.; Fan, Y.; Gao, Y.; Zhan, Y.; Wang, L.; Xiao, B.; Qiu, W.; Gu, X.; Sun, K.; et al. A 25 Mainland Chinese cohort of patients with PURA-related neurodevelopmental disorders: Clinical delineation and genotype-phenotype correlations. Eur. J. Hum. Genet. 2022, 31, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.; Leventer, R.J.; Simons, C.; Taft, R.; Swoboda, K.J.; Gawne-Cain, M.; DDD Study; Magee, A.C.; Turnpenny, P.D.; Baralle, D. Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J. Med. Genet. 2014, 51, 806–813. [Google Scholar] [CrossRef] [Green Version]
- Shimojima, K.; Isidor, B.; Le Caignec, C.; Kondo, A.; Sakata, S.; Ohno, K.; Yamamoto, T. A new microdeletion syndrome of 5q31.3 characterized by severe developmental delays, distinctive facial features, and delayed myelination. Am. J. Med. Genet. A 2011, 155A, 732–736, Erratum in Am. J. Med. Genet. A 2011, 155A, 2903. [Google Scholar] [CrossRef]
- Available online: https://www.purasyndrome.org/condition (accessed on 29 November 2022).
- Daigle, J.G.; Krishnamurthy, K.; Ramesh, N.; Casci, I.; Monaghan, J.; McAvoy, K.; Godfrey, E.W.; Daniel, D.C.; Johnson, E.M.; Monahan, Z.; et al. Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol. 2016, 131, 605–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, K.; Del Valle, L.; Muralidharan, V.; Gault, W.J.; Darbinian, N.; Otte, J.; Meier, E.; Johnson, E.M.; Daniel, D.C.; Kinoshita, Y.; et al. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol. Cell Biol. 2003, 23, 6857–6875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hokkanen, S.; Feldmann, H.M.; Ding, H.; Jung, C.K.; Bojarski, L.; Renner-Müller, I.; Schüller, U.; Kretzschmar, H.; Wolf, E.; Herms, J. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum. Mol. Genet. 2012, 21, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbe, M.F.; Krueger, J.J.; Loomis, R.; Otte, J.; Gordon, J. Memory deficits, gait ataxia and neuronal loss in the hippocampus and cerebellum in mice that are heterozygous for Pur-alpha. Neuroscience 2016, 337, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balice-Gordon, R.J.; Lichtman, J.W. In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J. Neurosci. 1993, 13, 834–855. [Google Scholar] [CrossRef] [PubMed]
- Dobretsova, A.; Johnson, J.W.; Jones, R.C.; Edmondson, R.D.; Wight, P.A. Proteomic analysis of nuclear factors binding to an intronic enhancer in the myelin proteolipid protein gene. J. Neurochem. 2008, 105, 1979–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, S.; Thatikunta, P.; Steplewski, A.; Johnson, E.M.; Khalili, K.; Amini, S. A 39-kD DNA-binding protein from mouse brain stimulates transcription of myelin basic protein gene in oligodendrocytic cells. J. Cell Biol. 1995, 130, 1171–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, Y.; Dohmae, N.; Hirokawa, N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron 2004, 43, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Mitsumori, K.; Takei, Y.; Hirokawa, N. Components of RNA granules affect their localization and dynamics in neuronal dendrites. Mol. Biol. Cell 2017, 28, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Kiebler, M.A.; Bassell, G.J. Neuronal RNA granules: Movers and makers. Neuron 2006, 51, 685–690. [Google Scholar] [CrossRef] [Green Version]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikbaev, A.; Manahan-Vaughan, D. Metabotropic glutamate receptor, mGlu5, regulates hippocampal synaptic plasticity and is required for tetanisation-triggered changes in theta and gamma oscillations. Neuropharmacology 2017, 115, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piers, T.M.; Kim, D.H.; Kim, B.C.; Regan, P.; Whitcomb, D.J.; Cho, K. Translational Concepts of mGluR5 in Synaptic Diseases of the Brain. Front. Pharmacol. 2012, 3, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezon-Geyda, K.; Najfeld, V.; Johnson, E.M. Deletions of PURA, at 5q31, and PURB, at 7p13, in myelodysplastic syndrome and progression to acute myelogenous leukemia. Leukemia 2001, 15, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Di Salvio, M.; Piccinni, V.; Gerbino, V.; Mantoni, F.; Camerini, S.; Lenzi, J.; Rosa, A.; Chellini, L.; Loreni, F.; Carrì, M.T.; et al. Pur-alpha functionally interacts with FUS carrying ALS-associated mutations. Cell Death Dis. 2015, 6, e1943. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.M.; Kinoshita, Y.; Weinreb, D.B.; Wortman, M.J.; Simon, R.; Khalili, K.; Winckler, B.; Gordon, J. Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J. Neurosci. Res. 2006, 83, 929–943. [Google Scholar] [CrossRef]
- Mizielinska, S.; Lashley, T.; Norona, F.E.; Clayton, E.L.; Ridler, C.E.; Fratta, P.; Isaacs, A.M. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013, 126, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Poidevin, M.; Li, X.; Li, Y.; Shu, L.; Nelson, D.L.; Li, H.; Hales, C.M.; Gearing, M.; Wingo, T.S.; et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 7778–7783. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, B.; Robberecht, W.; Van Den Bosch, L. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2020, 39, e101112. [Google Scholar] [CrossRef]
- Hariharan, S.; Kelm, R.J., Jr.; Strauch, A.R. The Purα/Purβ single-strand DNA-binding proteins attenuate smooth-muscle actin gene transactivation in myofibroblasts. J. Cell Physiol. 2014, 229, 1256–1271. [Google Scholar] [CrossRef]
- Pandey, P.R.; Yang, J.H.; Tsitsipatis, D.; Panda, A.C.; Noh, J.H.; Kim, K.M.; Munk, R.; Nicholson, T.; Hanniford, D.; Argibay, D.; et al. circSamd4 represses myogenic transcriptional activity of PUR proteins. Nucleic Acids Res. 2020, 48, 3789–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, K.; Reeve, A.; Bowes, M.; Winter, J.; Wotherspoon, G.; Davis, A.; Schmid, P.; Gasparini, F.; Kuhn, R.; Urban, L. mGlu5 receptors and nociceptive function II. mGlu5 receptors functionally expressed on peripheral sensory neurones mediate inflammatory hyperalgesia. Neuropharmacology 2001, 40, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Wyrebek, R.; Bartolomeo, M.D.; Brooks, S.; Geller, T.; Crenshaw, M.; Iyadurai, S. Corrigendum to ‘Hypotonic Infant with PURA Syndrome-related Channelopathy Successfully Treated with Pyridostigmine’ Neuromuscular Disorders Volume 32, Issue 2, February 2022, Pages 166-169. Neuromuscul. Disord. 2022, 32, e1, Erratum in Neuromuscul. Disord. 2022, 32, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Johannesen, K.M.; Gardella, E.; Gjerulfsen, C.E.; Bayat, A.; Rouhl, R.P.W.; Reijnders, M.; Whalen, S.; Keren, B.; Buratti, J.; Courtin, T.; et al. PURA-Related Developmental and Epileptic Encephalopathy: Phenotypic and Genotypic Spectrum. Neurol. Genet. 2021, 7, e613. [Google Scholar] [CrossRef]
- Lee, B.H.; Reijnders, M.R.F.; Abubakare, O.; Tuttle, E.; Lape, B.; Minks, K.Q.; Stodgell, C.; Bennetto, L.; Kwon, J.; Fong, C.T.; et al. Expanding the neurodevelopmental phenotype of PURA syndrome. Am. J. Med. Genet. A 2018, 176, 56–67. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guide—669 line for reporting systematic reviews. Rev. Esp. Cardiol. (Engl. Ed.) 2021, 74, 790–799, Erratum in Rev. Esp. Cardiol. (Engl. Ed.) 2022, 75, 192. [Google Scholar] [CrossRef]
- Qashqari, H.; McNiven, V.; Gonorazky, H.; Mendoza-Londono, R.; Hassan, A.; Kulkarni, T.; Amburgey, K.; Dowling, J.J. PURA syndrome: Neuromuscular junction manifestations with potential therapeutic implications. Neuromuscul. Disord. 2022, 32, 842–844. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Janowski, R.; Alvi, M.; Self, J.E.; van Essen, T.J.; Vreeburg, M.; Rouhl, R.P.W.; Stevens, S.J.C.; Stegmann, A.P.A.; Schieving, J.; et al. PURA syndrome: Clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J. Med. Genet. 2018, 55, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.; Burgess, T.; Forbes, R.; McGillivray, G.; Kornberg, A.; Mandelstam, S.; Stark, Z. 5q31.3 Microdeletion syndrome: Clinical and molecular characterization of two further cases. Am. J. Med. Genet. A 2013, 161A, 2604–2608. [Google Scholar] [CrossRef]
- Bonaglia, M.C.; Zanotta, N.; Giorda, R.; D’Angelo, G.; Zucca, C. Long-term follow-up of a patient with 5q31.3 microdeletion syndrome and the smallest de novo 5q31.2q31.3 deletion involving PURA. Mol. Cytogenet. 2015, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.J.; Bai, R.; Cho, M.T.; Anyane-Yeboa, K.; Ahimaz, P.; Wilson, A.L.; Kendall, F.; Hay, B.; Moss, T.; Nardini, M.; et al. De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000356. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, N.; Nakao, H.; Niihori, T.; Aoki, Y. Patient with a novel purine-rich element binding protein A mutation. Congenit. Anom. 2017, 57, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Rezkalla, J.; Von Wald, T.; Hansen, K.A. Premature Thelarche and the PURA Syndrome. Obstet. Gynecol. 2017, 129, 1037–1039. [Google Scholar] [CrossRef]
- Mayorga, L.; Gamboni, B.; Mampel, A.; Roqué, M. A frame-shift deletion in the PURA gene associates with a new clinical finding: Hypoglycorrhachia. Is GLUT1 a new PURA target? Mol. Genet. Metab. 2018, 123, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, K.; Okamoto, N.; Ohmura, K.; Nagase, H.; Yamamoto, T. Infantile spasms related to a 5q31.2-q31.3 microdeletion including PURA. Hum. Genome Var. 2018, 5, 18007. [Google Scholar] [CrossRef] [Green Version]
- Trau, S.P.; Pizoli, C.E. PURA Syndrome and Myotonia. Pediatr. Neurol. 2020, 104, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-García, M.E.; Cotrina-Vinagre, F.J.; Arranz-Canales, E.; Aragón, A.M.; Hernández-Sánchez, L.; Rodríguez-Fornés, F.; Carnicero-Rodríguez, P.; Morales-Conejo, M.; Martín-Hernández, E.; Martínez-Azorín, F. A novel de novo mutation in the PURA gene associated with a new clinical finding: Large brainstem. J. Genet. 2020, 99, 7. [Google Scholar] [CrossRef] [PubMed]
- Jezela-Stanek, A.; Ciara, E.; Jurkiewicz, D.; Kucharczyk, M.; Jędrzejowska, M.; Chrzanowska, K.H.; Krajewska-Walasek, M.; Żemojtel, T. The phenotype-driven computational analysis yields clinical diagnosis for patients with atypical manifestations of known intellectual disability syndromes. Mol. Genet. Genom. Med. 2020, 8, e1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinquina, V.; Ciaccio, C.; Venturini, M.; Masson, R.; Ritelli, M.; Colombi, M. Expanding the PURA syndrome phenotype: A child with the recurrent PURA p. (Phe233del) pathogenic variant showing similarities with cutis laxa. Mol. Genet. Genom. Med. 2021, 9, e1562. [Google Scholar] [CrossRef]
- Mroczek, M.; Zafeiriou, D.; Gurgel-Gianetti, J.; Vilela Morais de Azevedo, B.; Roos, A.; Bartels, E.; Kohlschmidt, N.; Phadke, R.; Feng, L.; Duff, J.; et al. Three Individuals with PURA Syndrome in a Cohort of Patients with Neuromuscular Disease. Neuropediatrics 2021, 52, 390–393. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, R.; Xu, T.; Zhou, Y.X.; Zhang, S.C. Neonatal PURA syndrome: A case report and literature review. Transl. Pediatr. 2021, 10, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Solazzi, R.; Fiorini, E.; Parrini, E.; Guerrini, R.; Bernardina, B.D.; Nardocci, N.; Cantalupo, G. Early-onset bradykinetic rigid syndrome and reflex seizures in a child with PURA syndrome. Epileptic Disord. 2021, 23, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.A.; Lee, H.S.; Park, T.J.; Park, S.; Ko, Y.J.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Chae, J.H. Expanding the clinical phenotype and genetic spectrum of PURA-related neurodevelopmental disorders. Brain Dev. 2021, 43, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Lin, Y.F.; Tsai, C.H.; Huang, C.H.; Ho, F.; Tsai, S.F.; Lin, W.S. Complex Movement Disorders in a Boy with PURA Syndrome. Mov. Disord. Clin. Pract. 2021, 8, 1137–1139. [Google Scholar] [CrossRef]
- Nogueira, M.; Melo, C.; Grangeia, A.; Magalhães, T.; Soares, C.; Dias, R.; Fonseca, J.; Sampaio, M.; Sousa, R. PURA syndrome in a child with severe developmental delay: A challenging diagnosis. Rev. Neurol. 2022, 74, 170–173. [Google Scholar] [CrossRef]
- Fukuda, Y.; Kudo, Y.; Saito, M.; Kaname, T.; Oota, T.; Shoji, R. Expanding the PURA syndrome phenotype with manifestations in a Japanese female patient. Hum. Genome Var. 2022, 9, 11. [Google Scholar] [CrossRef]
- Lisi, E.C.; Cohn, R.D. Genetic evaluation of the pediatric patient with hypotonia: Perspective from a hypotonia specialty clinic and review of the literature. Dev. Med. Child Neurol. 2011, 53, 586–599. [Google Scholar] [CrossRef]
- Patel, J.C.; Barvaliya, M.J.; Patel, T.K.; Tripathi, C.B. Neuromuscular blocking effect of fluoxetine and its interaction with rocuronium. Auton. Autacoid. Pharmacol. 2013, 33, 17–24. [Google Scholar] [CrossRef]
- Bertrand, C.; Bonafos, B.; Tremblay, M.; Ferry, A.; Chatonnet, A. Effect of fluoxetine on neuromuscular function in acetylcholinesterase (AChE) knockout mice. Chem. Biol. Interact. 2008, 175, 113–114. [Google Scholar] [CrossRef]
- Engel, A.G. The therapy of congenital myasthenic syndromes. Neurotherapeutics 2007, 4, 252–257, Erratum in Neurotherapeutics 2019, 16, 244. [Google Scholar] [CrossRef] [Green Version]
- Hedrich, U.B.S.; Lauxmann, S.; Wolff, M.; Synofzik, M.; Bast, T.; Binelli, A.; Serratosa, J.M.; Martínez-Ulloa, P.; Allen, N.M.; King, M.D.; et al. 4-Aminopyridine is a promising treatment option for patients with gain-of-function KCNA2-encephalopathy. Sci. Transl. Med. 2021, 13, eaaz4957. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Yes | No | % |
|---|---|---|
| Hypotonia | ||
| 187 | 4 | 98 |
| Respiratory | ||
| 124 | 20 | 86% |
| Myopathic face | ||
| 53 | 32 | 63% |
| Ambulation | ||
| 73 | 79 | 48% |
| Ptosis | ||
| 7 | 39 | 15% |
| DTR | ||
| Abnormal | Normal | |
| 17 | 3 | |
| EMG/NCV | ||
| Abnormal | Normal | |
| 9 | 7 | |
| Muscle biopsy | ||
| Abnormal | Normal | |
| 9 | 3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mroczek, M.; Iyadurai, S. Neuromuscular and Neuromuscular Junction Manifestations of the PURA-NDD: A Systematic Review of the Reported Symptoms and Potential Treatment Options. Int. J. Mol. Sci. 2023, 24, 2260. https://doi.org/10.3390/ijms24032260
Mroczek M, Iyadurai S. Neuromuscular and Neuromuscular Junction Manifestations of the PURA-NDD: A Systematic Review of the Reported Symptoms and Potential Treatment Options. International Journal of Molecular Sciences. 2023; 24(3):2260. https://doi.org/10.3390/ijms24032260
Chicago/Turabian StyleMroczek, Magdalena, and Stanley Iyadurai. 2023. "Neuromuscular and Neuromuscular Junction Manifestations of the PURA-NDD: A Systematic Review of the Reported Symptoms and Potential Treatment Options" International Journal of Molecular Sciences 24, no. 3: 2260. https://doi.org/10.3390/ijms24032260
APA StyleMroczek, M., & Iyadurai, S. (2023). Neuromuscular and Neuromuscular Junction Manifestations of the PURA-NDD: A Systematic Review of the Reported Symptoms and Potential Treatment Options. International Journal of Molecular Sciences, 24(3), 2260. https://doi.org/10.3390/ijms24032260